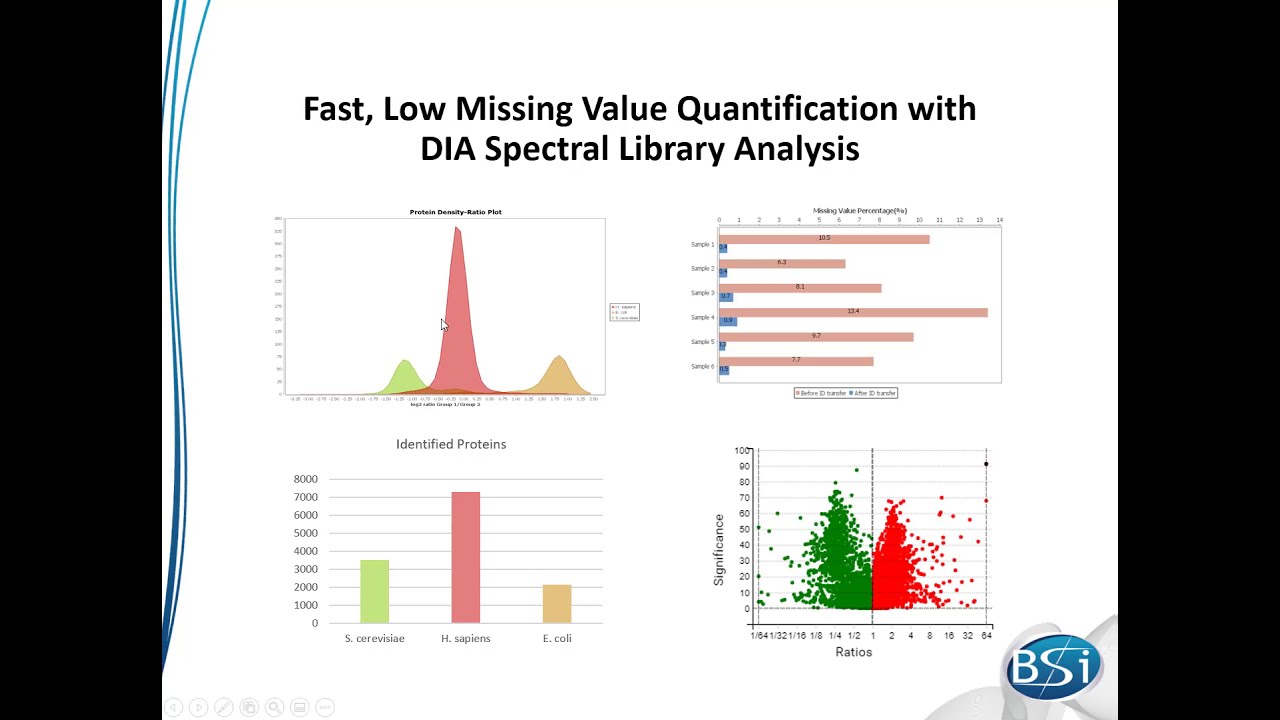
0On 2019-10-302023-03-30By Bioinformatics Solutions Inc. This video introduces the major new features provided by the newly released PEAKS X+. PEAKS Studio X+ brings some exciting new features to the PEAKS Studio protein identification and quantification platform. This update brings: Peptide spectral library creation Peptide spectral library search Retention time prediction assisted de novo sequencing Quality control statistics – Identification led label-free quantification (LFQ) Click >>HERE<< to download PEAKS X+ and request a trial key. New in PEAKS Studio 809 views You may also like PEAKS Online Xpro Walkthrough 1,143 views New in PEAKS Studio PEAKS Studio Xpro Walkthrough 753 views New in PEAKS Studio PEAKS Xpro Overview 627 views New in PEAKS Studio PEAKS X Introduction Video 1,327 views New in PEAKS Studio PEAKS Q Labelling Quantification – SILAC 1,351 views New in PEAKS Studio PEAKS Studio 8.5 Overview of Features 755 views New in PEAKS Studio




Leave a Reply
You must be logged in to post a comment.