Featured Article:
Abstract
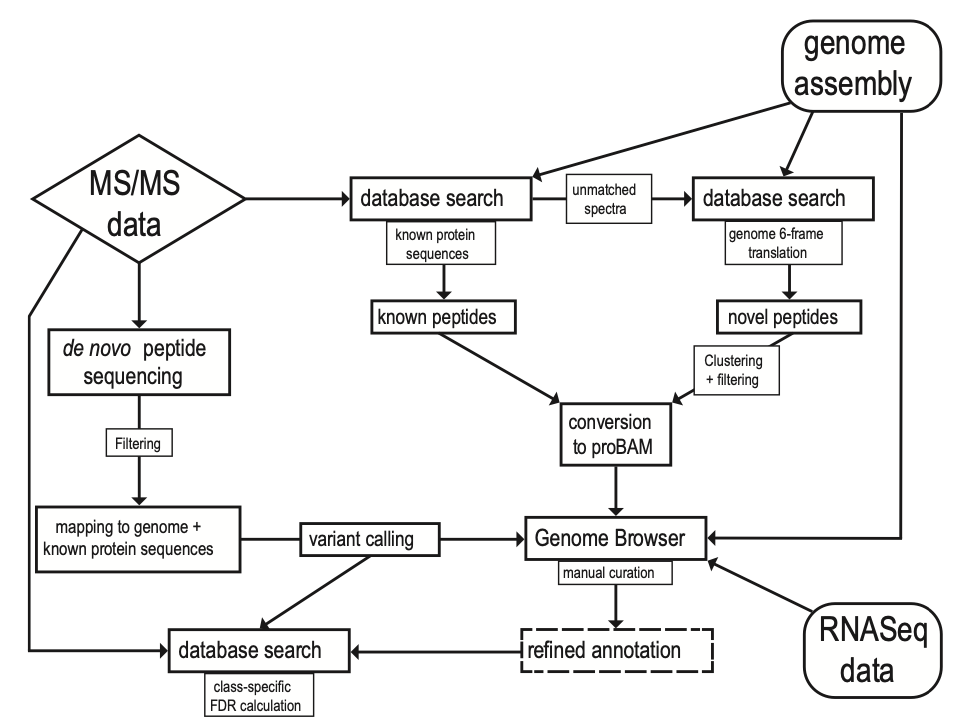
A recent publication by Blank-Landeshammer et al. in the American Society for Microbiology mBio journal highlights the power of combining proteogenomic tools with PEAKS de novo sequencing to identify new genes and hidden posttranscriptional modifications. Since not all proteins can be predicted from genetic sequences, there is a requirement for de novo peptide sequencing to reveal new functional components within proteomes of species across all domains of life. The field of proteogenomics has helped to refine genome annotations, however, this type of analysis does not identify peptides modified by posttranscriptional changes (i.e., RNA editing, alternative spliced variants, fissions, and fusions). In this article, the authors use de novo sequencing technology from PEAKS to help detect 104 hidden proteins from the well-studied model fungus Sordaria macrospora. Moreover, their analysis led to a re-annotation of 575 genes, including 389 splice site refinements, 113 single-amino-acid variations, and 15 C-terminal protein extensions caused by RNA editing (UAG to UGG stop codon loss). The de novo peptide sequencing function of PEAKS, along with scoring functions and retention time predictions, were employed to accurately identify novel peptides. This was a critical aspect of the analysis since PEAKS de novo sequencing does not rely on peptide databases, which are often derived from gene sequences predicted by a homology-based approach. While newly sequenced organisms would be well-suited for this type of study, the authors demonstrate that the combination of proteogenomics and de novo sequencing can significantly improve genome annotation quality of even well-studied organisms.