de novo Peptide Sequencing
Key Features:
- Integration of deep neural network model, DeepNovo, for peptide de novo sequencing to increase amino acid and peptide level accuracy
- Automated de novo peptide sequencing with high throughput
- Accuracy at amino acid level
- Combines complementary fragmentations
- Supports CID, HCD, ETD/ECD, EThcD, UVPD
- Integrates with database search

In a tandem mass spectrometer, the peptide is fragmented along the peptide backbone and the resulting fragment ions are measured to produce the MS/MS spectrum. Depending on the fragmentation method used, different fragment ion types can be produced. de novo Peptide sequencing derives an amino acid sequence from a mass spectrum without the need of a sequence database. It is in contrast to another popular peptide identification approach – “database search”, which searches in a given database to find the largest peptide. De novo peptide sequencing is the only choice when the sequence database is not available. This makes PEAKS the preferred method for identifying novel peptides and proteins from unsequenced organisms.
DeepNovo
DeepNovo is a deep learning based algorithm for de novo sequencing that predicts the peptide from the MS/MS scan by iteratively predicting amino acids consecutively. At each step, DeepNovo predicts the next amino acid and its score. The score represents the probability of a particular amino acid to be present at a position within the peptide sequence . The score of the predicted peptide is then calculated as the average of its amino acid scores. This new tool can increase amino acid level accuracy, and amino acid and peptide recall.
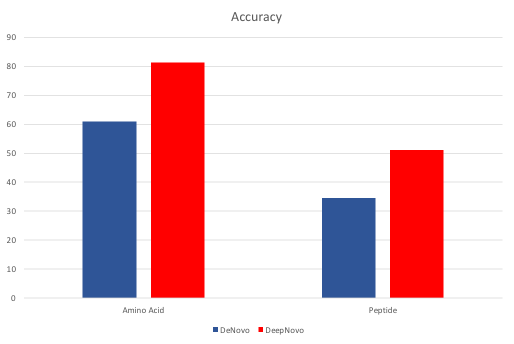
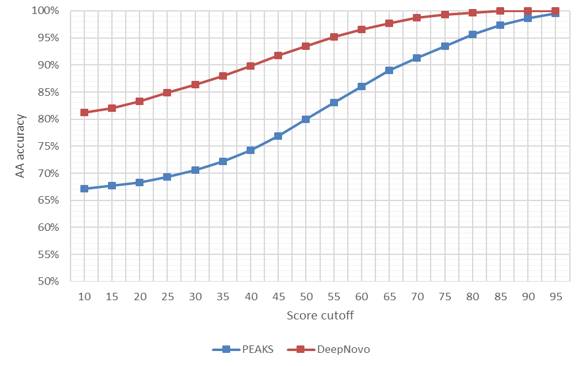
Accuracy of PEAKS de novo and DeepNovo predictions of PSMs at the amino acid and peptide levels from TimsTOF instrument and HLA dataset.
Accuracy
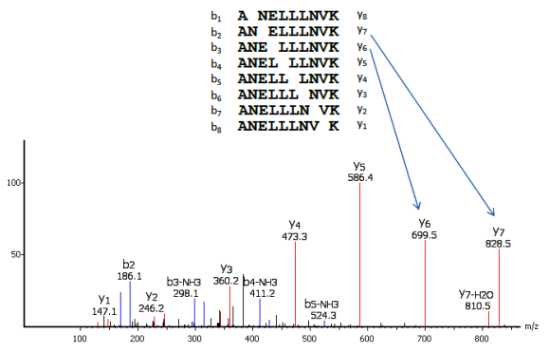
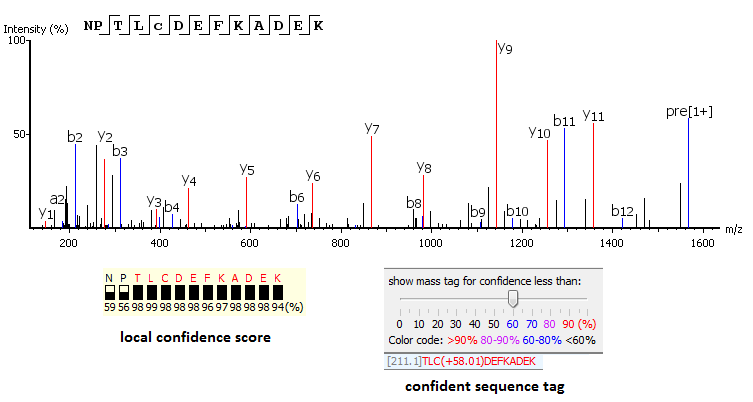
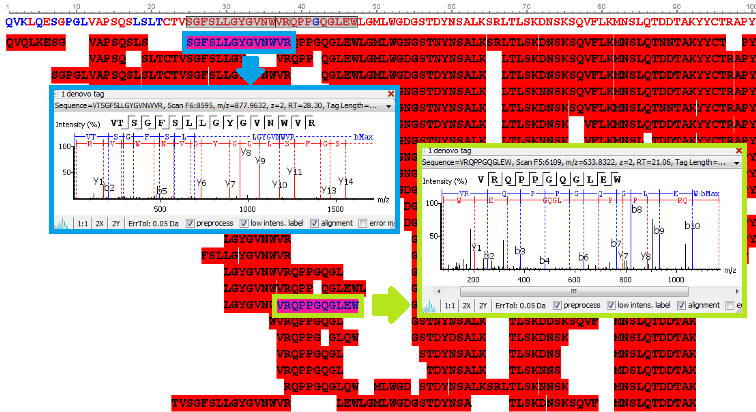
PEAKS uses a comprehensive scoring system to provide accurate de novo peptide sequencing results. Unique to PEAKS is the Local Confidence Score – the likelihood of each amino acid assignment in a resultant peptide. The local confidence score extends the accuracy to the amino acid level. In the figure, TLCDEFKADEK is a confident sequence tag with significant fragment proof.
Complementary Fragmentation
Utilisation of Spectrum-Pairs: de novo sequencing using spectrum-pairs that are generated in different fragmentation modes (eg. ETD/HCD). Confident de novo sequence tags from each complementary spectrum are used to reconstruct a peptide sequence, which is optimised to both spectra.
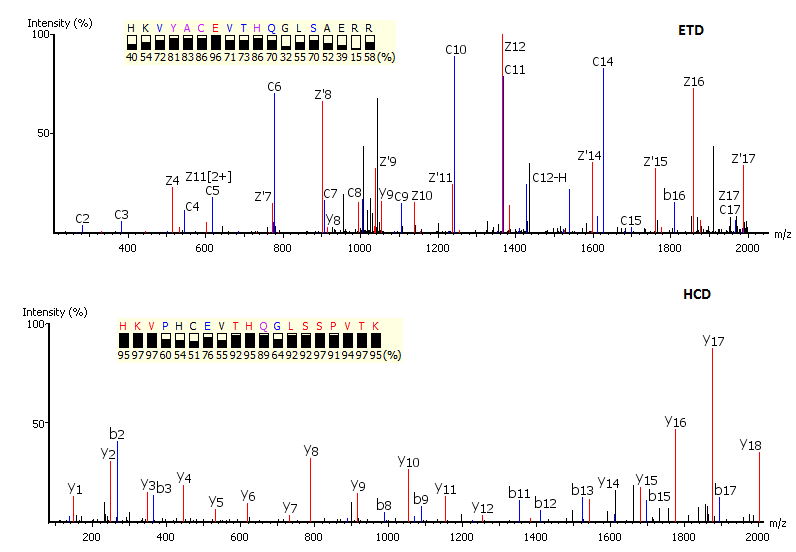
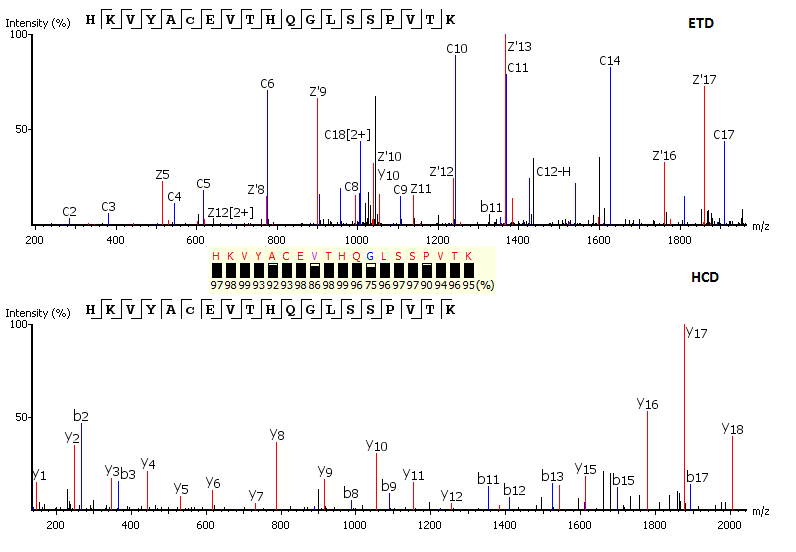
Integrated with Database Search
Unique to PEAKS is the ability to combine de novo peptide sequencing results with those of a database search. De novo peptide sequences are aligned with protein database entries to provide additional information about PTMs, mutations, homologous peptides, and novel peptides.
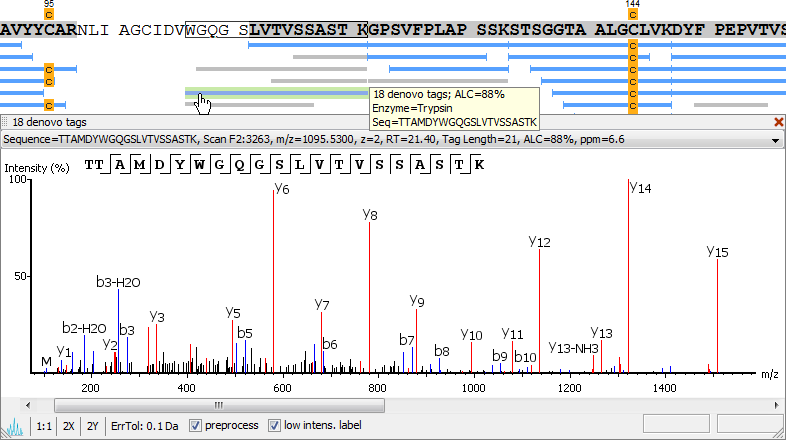
From de novo Peptide Sequencing to Protein Sequencing
Protein sequences could be obtained from the de novo peptide sequences. The confident de novo peptide sequence tags, which have direct fragmentation ion proof, were assembled into protein sequences, See PEAKS AB Software.
Reference
- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods. 16(1), 63-66. 20/12/2018.
- Tran NH, Zhang X, Xin L, Shan B, Li M. De novo peptide sequencing by deep learning. Proceedings of the National Academy of Sciences of the United States of America. 114(29). 18/7/2017.
- Tran NH, Rahman MZ, He L, Xin L, Shan B, Li M. Complete De Novo Assembly of Monoclonal Antibody Sequences Scientific Reports. 6(31730). 26/08/2016.
- He L, Ma B. ADEPTS: advanced peptide de novo sequencing with a pair of tandem mass spectra. Journal of Bioinformatics and Computational Biology. 8(06):981-994. 1/12/2012.
- Qiao R ,Tran NH ,Xin L ,Chen X , Li M , Shan B, and Ghodsi A, Computationally instrument-resolutionindependent de novo peptide sequencing for high-resolution devices, Nat. Mach. Intell, 3, 420–425 (2021). doi:10.1038/s42256-021-00304-3