
 Database Search Scoring
Database Search Scoring
Understanding the fundamentals
LDF Score
The score calculation in PEAKS DB can be illustrated in the following figure.
PEAKS DB internally uses an LDF (linear discriminative function) score to measure the quality of the peptide-spectrum match. The LDF score uses not only the matching between the fragment ions and the peaks in the spectrum, but also many other factors such as the similarity between the de novo sequencing peptide and the database peptide.
This LDF score tries to achieve two goals:
- for each spectrum in the MS/MS data set, finds the most likely correct peptide from the database;
- for the whole data set, separates the true and false identifications as much as possible.
P-Value
The LDF score is converted to a P-value for better human interpretation.
P-value: for a given score x, its equivalent P-value is the probability that a false identification has score “>” x.
The smaller the P-value is, the less likely the peptide-spectrum match is due to a random match. The meaning of the P-value is better explained in the following figure.
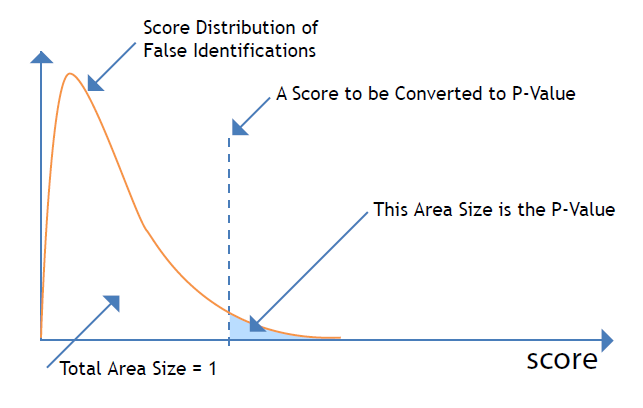
Note that although many software packages use the same term “P-value”, their meanings can be different. Another popular definition of P-value is “the probability as that a random peptide matches the current spectrum with score “>” x”. However, in the database search, a false identification is the result of many random peptides in the database, instead of just one random peptide. Thus, the definition in PEAKS DB is more useful for the controlling of result quality with P-value.
-10logP
The P-value is converted to -10*log10(P-value) to make it more “human-friendly”. In PEAKS, this value is denoted by -10lgP as lg is the ISO reserved notation for log10. By this conversion, a more significant match will have a higher -10lgP value. Additionally, a P-value of 1% is equivalent to -10lgP of 20.
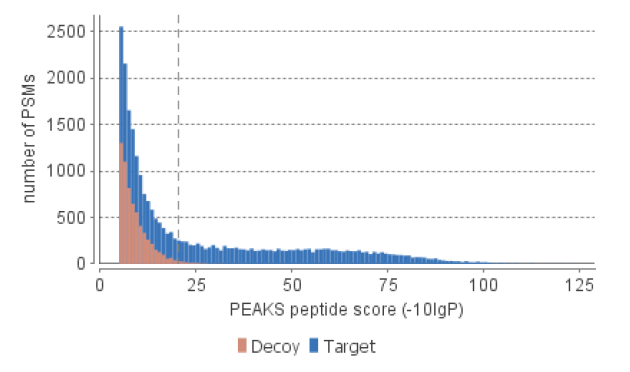
The following figure is a screenshot from a PEAKS DB search result. The x-axis is the -10lgP score, and the y-axis is the number of peptide-spectrum matches with that score. Normally, a greater than 20 score is of relatively high confidence (as illustrated by many target but very few decoy matches above that threshold). For a large dataset, it is recommended to use the FDR (false discovery rate) to select the right -10lgP score threshold (which is easy in PEAKS). However, when the dataset is small (# spectra “<” 100 or protein database contains only a few proteins), 20 is a good threshold for -10lgP.
References
- Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, Zhang W, Zhang Z, Lajoie G.A., Ma B, PEAKS DB: De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Mol. Cell. Proteomics. 11, M111.010587 (2012). https://doi.org/10.1074/mcp.M111.010587