

PEAKS AB 3.0
Protein de novo sequencing and glycan profiling software
Key Features
- Automated protein de novo sequencing
- Sequence non-antibody proteins with reference sequence
- In-depth glycan profiling
- Enhanced Ile/Leu differentiation
- Quantification of modifications and variants
- QC/QA of antibody production
- Utilise your expertise with manual editing
- Comprehensive report generation

Contact us to add PEAKS AB 3.0 to your lab!
The newest release of PEAKS AB Software, version 3.0, is equipped with new tools to further improve sequencing accuracy. It also provides a more convenient and user-friendly workflow for antibody sequencing analysis with new customisation capabilities.
Monoclonal antibodies have a wide range of applications in biochemical analyses, medical research, diagnostics and therapeutics. The protein sequence of an antibody (Ab) encodes critical information for its structure and function. When cDNA or the hybridoma cell line is not available, de novo protein sequencing of an antibody can help.
PEAKS AB Software is a platform for de novo antibody sequencing and the characterisation of modifications and sequence variants by using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). PEAKS AB software also supports characterisation of post-translational modifications (PTMs) and variants for research discovery and quality control purposes.
Software Overview
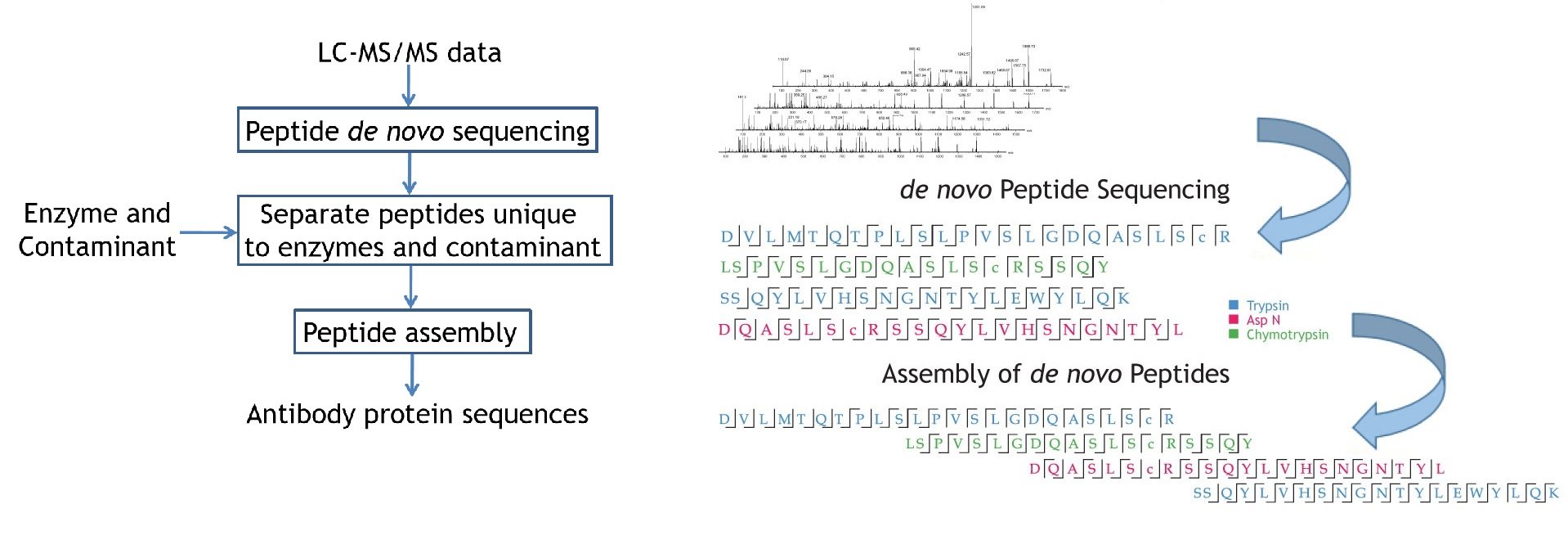
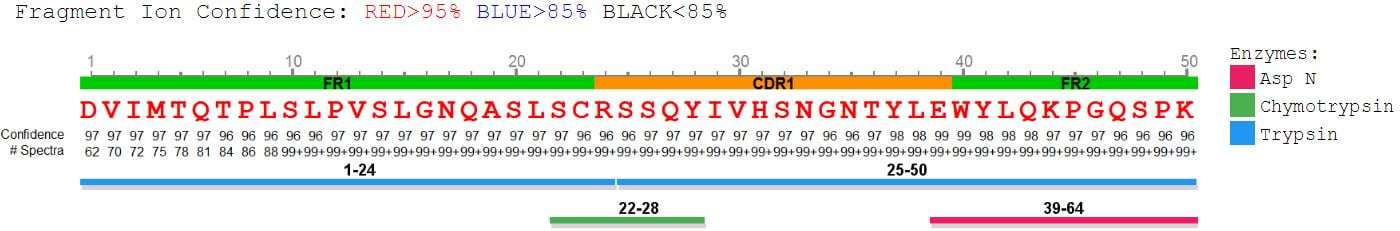
Antibody protein sequences were obtained from the antibody de novo peptide sequences. The confident de novo sequence tags, which have direct fragment ion proof, were assembled into antibody sequences. The de novo View shows the map of de novo peptide sequence tags.
de novo View
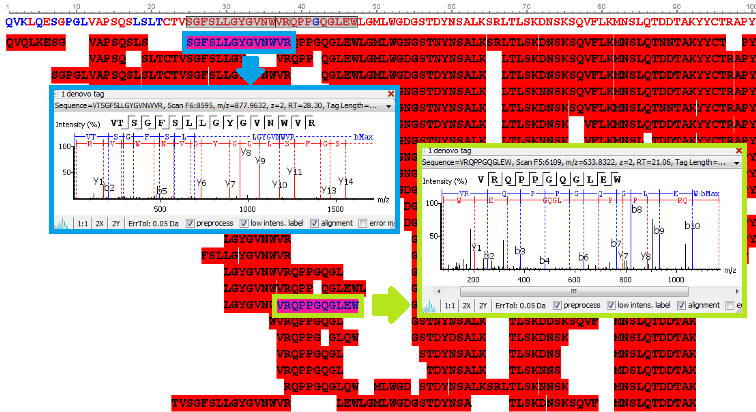
PEAKS AB provides comprehensive peptide maps, including:
- insight into full sequence information
- displaying each amino acid with confidence from direct fragment ions
- post translational modifications (PTMs)
- amino acid substitutions (sequence variants)
Peptide Mapping View
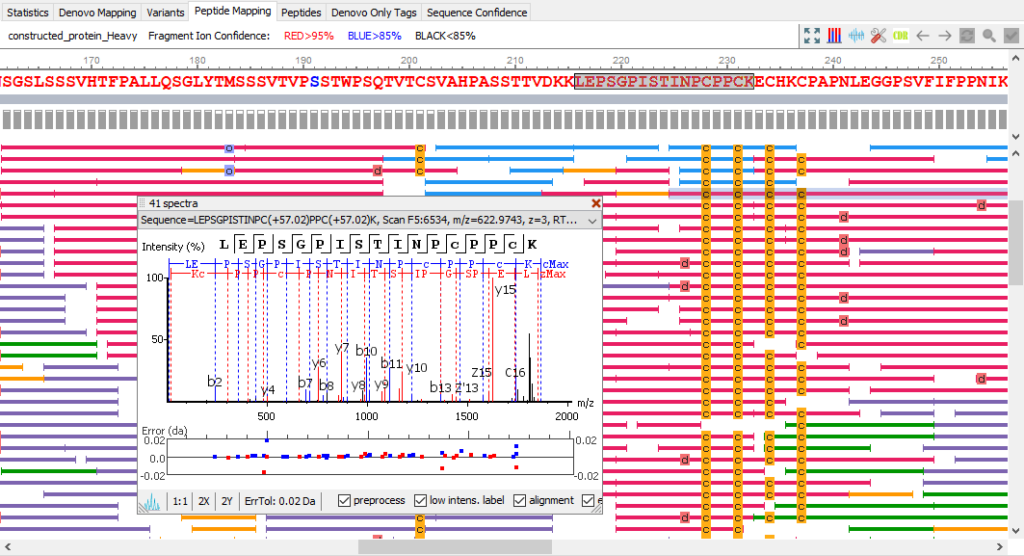
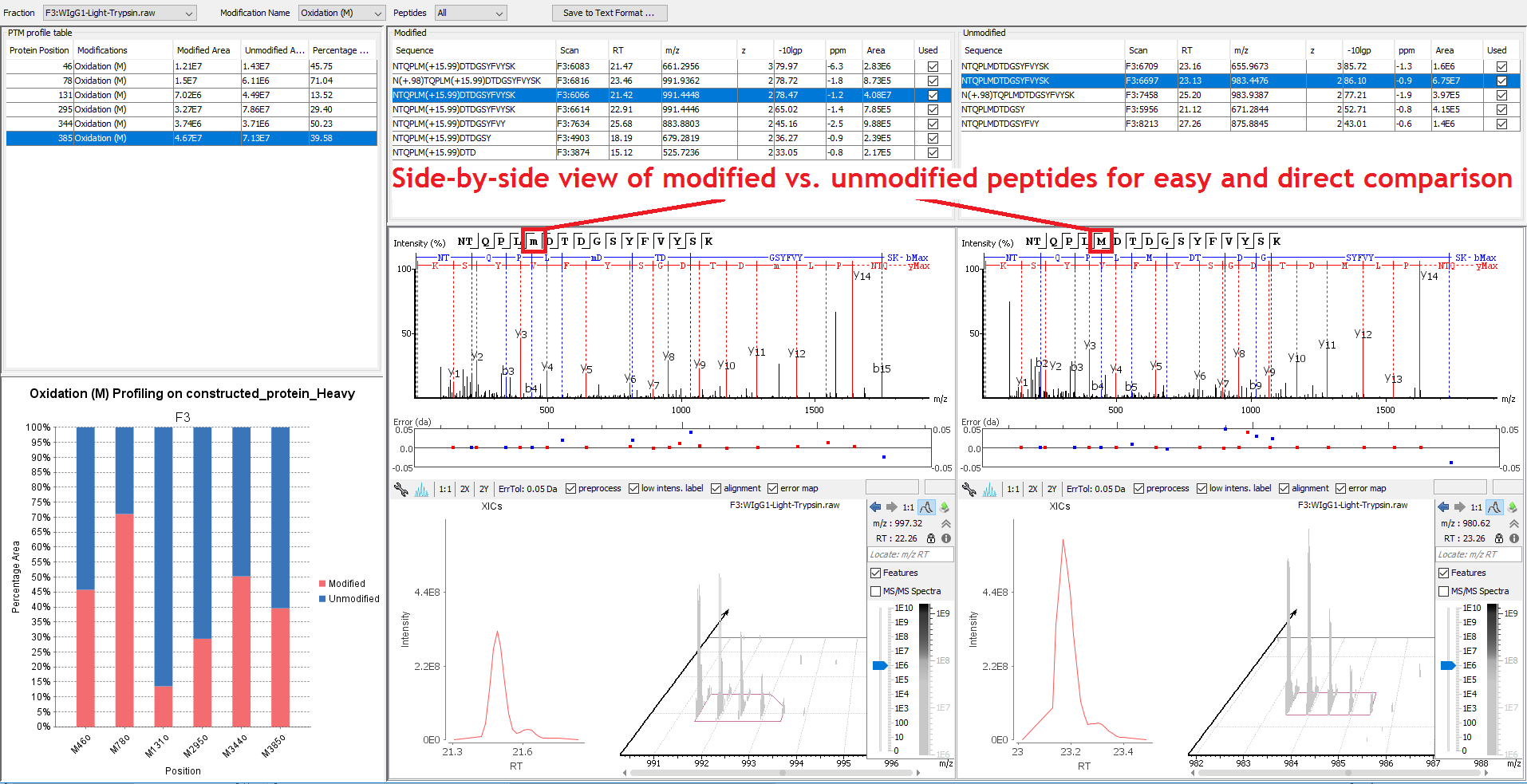
For each sequence position with modifications, the modified and native peptide forms, along with their chromatogram are listed for quantitative viewing.
Post-translational Modifications (PTMs) Analysis
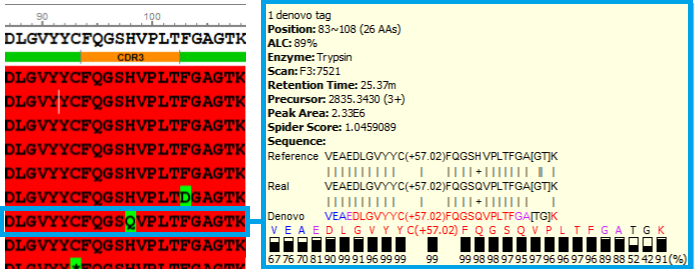
Sequence Variant View highlights amino acid substitutions and/or truncations which may result from erroneous transcription of complementary DNA.
Sequence Variant View
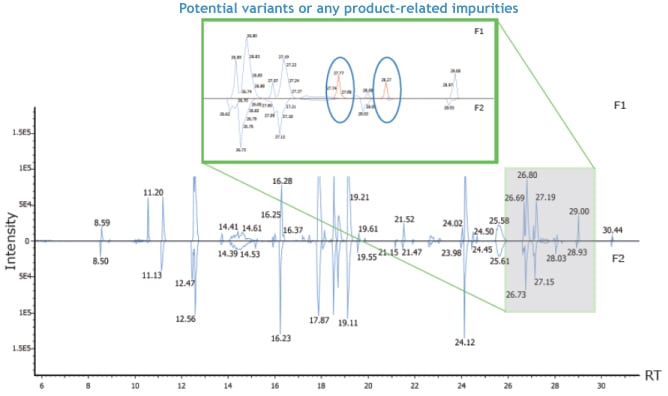
With the enhanced “Peptide Mapping” function, PEAKS AB Software can compare annotated chromatograms between different samples. Possible sequence variants will be illustrated to facilitate the product quality control or any other related antibody studies.
As shown in the image to the right, blue peaks in the annotated chromatogram denote the signals that can be mapped to peptides from reference proteins, and the red peaks denote sequence variants or any product-related impurities.
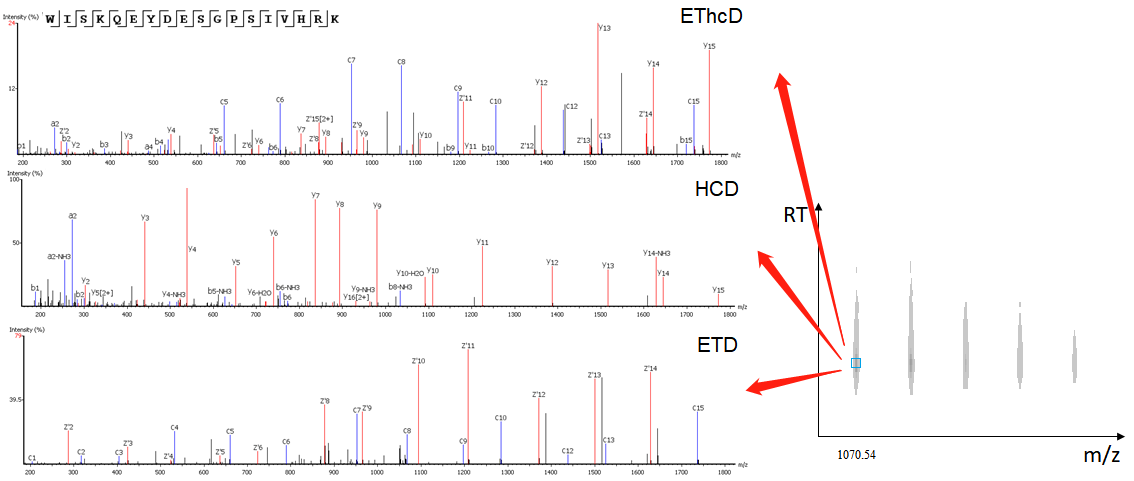
The EThcD fragmentation model was implemented in PEAKS AB 2.0. Moreover, MS2 spectra derived from the same precursor with different types of fragmentations are combined for de novo sequencing, yielding more confident de novo sequencing results.
EThcD Data Analysis
Algorithm
PEAKS AB software directly sequences the antibody protein using high resolution LC-MS/MS data sets from orthogonal enzyme digests. It performs de novo peptide sequencing first, and assembles the confident sequence tags into antibody sequences.
Report Generation
- Antibody Sample Description
- Abbreviations
- Experimental Procedure
- Antibody Heavy Chain
- Amino Acid Sequence
- Peptide Mapping at 0.1% of FDR at Peptide-Spectrum Level
- Typical Peptides Selected for Variable Region
- Ile/Leu Differentiation
- Antibody Light Chain
- Amino Acid Sequence
- Peptide Mapping at 0.1% of FDR at Peptide-Spectrum Level
- Typical Peptides Selected for Variable Region
- Ile/Leu Differentiation
- Glycan data can be reported using “Generate report” button in Glycan Profiling node.
References & Resources
References
- Shan, B. and Xin, L. Integrating de novo Sequencing and Database Search for Monoclonal Antibody Sequencing. J Biomol Tech. 24(Suppl). S62–S63. 1/5/2013.
- Tran NH, Rahman MZ, He L, Xin L, Shan B, Li M. Complete De Novo Assembly of Monoclonal Antibody Sequences. Sci. Rep., 6, 31730 (2016). https://doi.org/10.1038/srep31730