PEAKS DIA Workflow

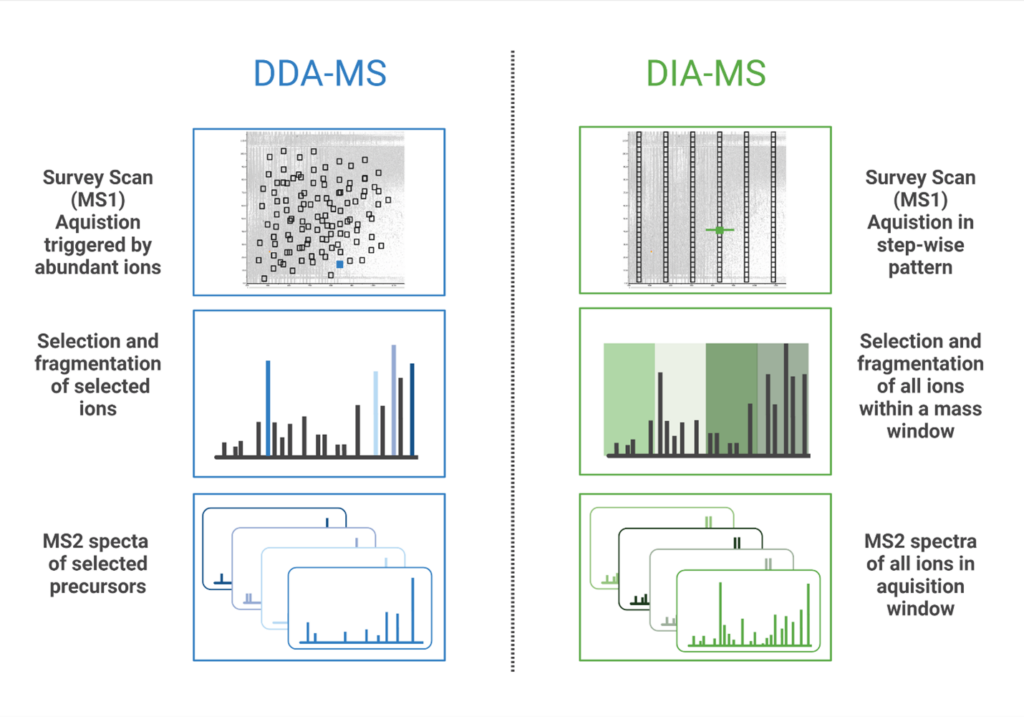
Data Independent Acquisition (DIA) is a mass spectrometry (MS) method where all ions within a defined mass-to-change (m/z) window are fragmented and analysed. Contrary to Data Dependent Acquisition (DDA), DIA workflows can reproducibly identify and quantify peptides without being limited to the most abundant precursor ions. In DDA mode, the mass spectrometer selects the most intense precursor ions from the MS1 scan, and only these selected peptides are fragmented for tandem MS analysis. In DIA, MS/MS spectra are acquired by either fragmenting all ions that enter the mass spectrometer at a given time (broadband DIA) or by sequentially acquiring ions within fixed or variable m/z windows for fragmentation (SWATH (Sequential Windowed Acquisition of All Theoretical Fragment ions)(1)). More recently, diaPASEF (parallel accumulation-serial fragmentation) technology was developed that uses Trapped Ion Mobility Separation (TIMS) with DIA to improve sensitivity of peptide detection and increase the depth of proteome coverage (2). Both PEAKS Studio & Online accommodates all types of DIA data mentioned above, to facilitate your analysis regardless of which method is used.
Advantages of a DIA Workflow
- Decrease bias by including all peptides in analysis
- Reproducibility of peptide detection and quantification across MS runs (3)
- Quantify proteins in complex mixtures over a dynamic range
- Eliminate under sampling
- Increased sensitivity and depth of proteome coverage
- Increased precision and reproducibility when compared to DDA (4)
- Eliminate the cost and time with label free quantification
PEAKS 12 New Features
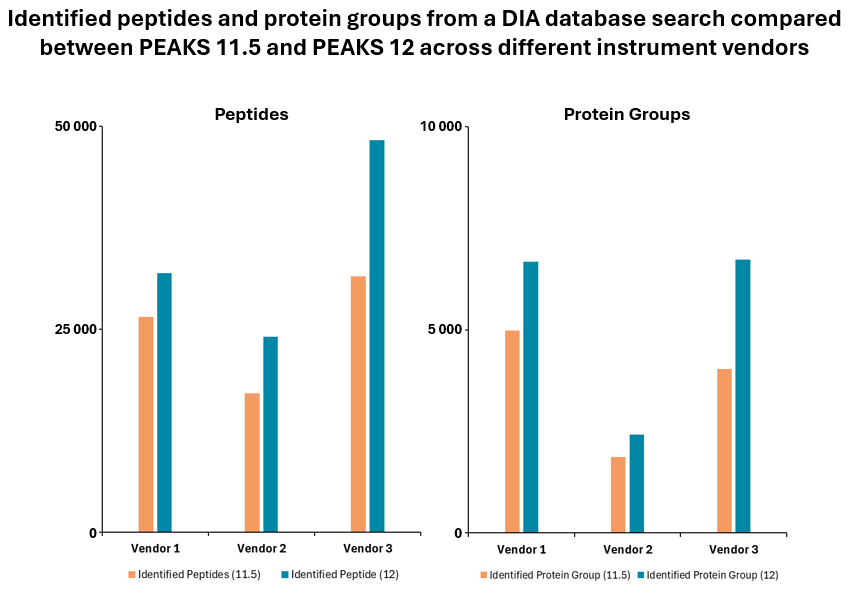
- Improved algorithm with up to 20% increase in peptide and protein identifications
- Complete solution for discovery and targeted proteomics in one software package
- DeepNovo Peptidome workflow for DIA
- Optional Match Between Runs step to increase coverage
- Support for MSFragger spectral library format
NEW PEAKS DIA Workflow
Maximise your identification rate from DIA datasets using the PEAKS DIA workflow
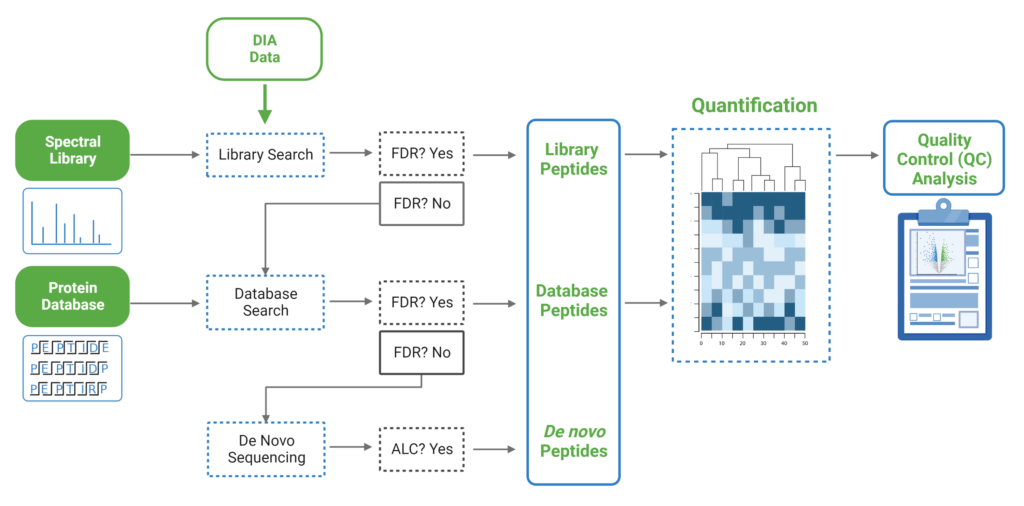
Since DIA MS/MS spectra are produced from a range of precursor ions within an m/z window, they are often multiplexed (contain signal from multiple peptides). This creates a unique challenge for an algorithm to identify peptides from DIA spectra. PEAKS 12 offers a powerful and flexible solution to this problem by allowing for peptide identification and quantification using spectral library search with or without a protein inference database, direct database search, or a combination of both for optimal peptide coverage (5). In addition, de novo sequencing can be applied to ensure no good spectra remain unidentified.
For a combined library and direct database search, the search is performed using an expanding search space. First, a library search is performed against a library of previously identified spectra. By calculating the false discovery rate, peptides that pass the filter are saved. MS/MS spectra that don’t match a peptide within the false discovery rate threshold are brought forward to a direct database search. Confident database matches are added to the result. Then, using the same FDR approach, unmatched spectra from the database search are analysed using de novo sequencing.
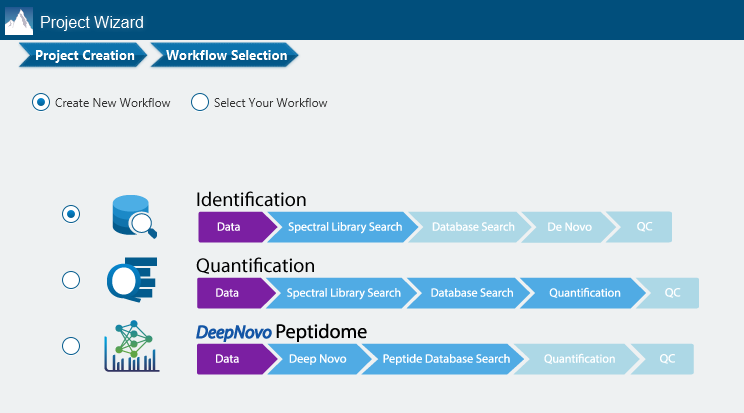
There are several workflows available for DIA data. Traditionally, the suggested workflow is to generate a library from DDA data and use this library when searching DIA data. A subsequent database search can be done to identify more peptides. Alternatively, a direct database search with DIA data (without a prior spectral library search) can also be done with PEAKS 12. Additionally, remaining unmatched scans can be de novo sequenced. For quantification, the label-free method is available as an add-on module. Lastly, in PEAKS 12 the QC analysis offers statistical summary and visualisation of data quality control measures for both Identification and Quantification. Learn more
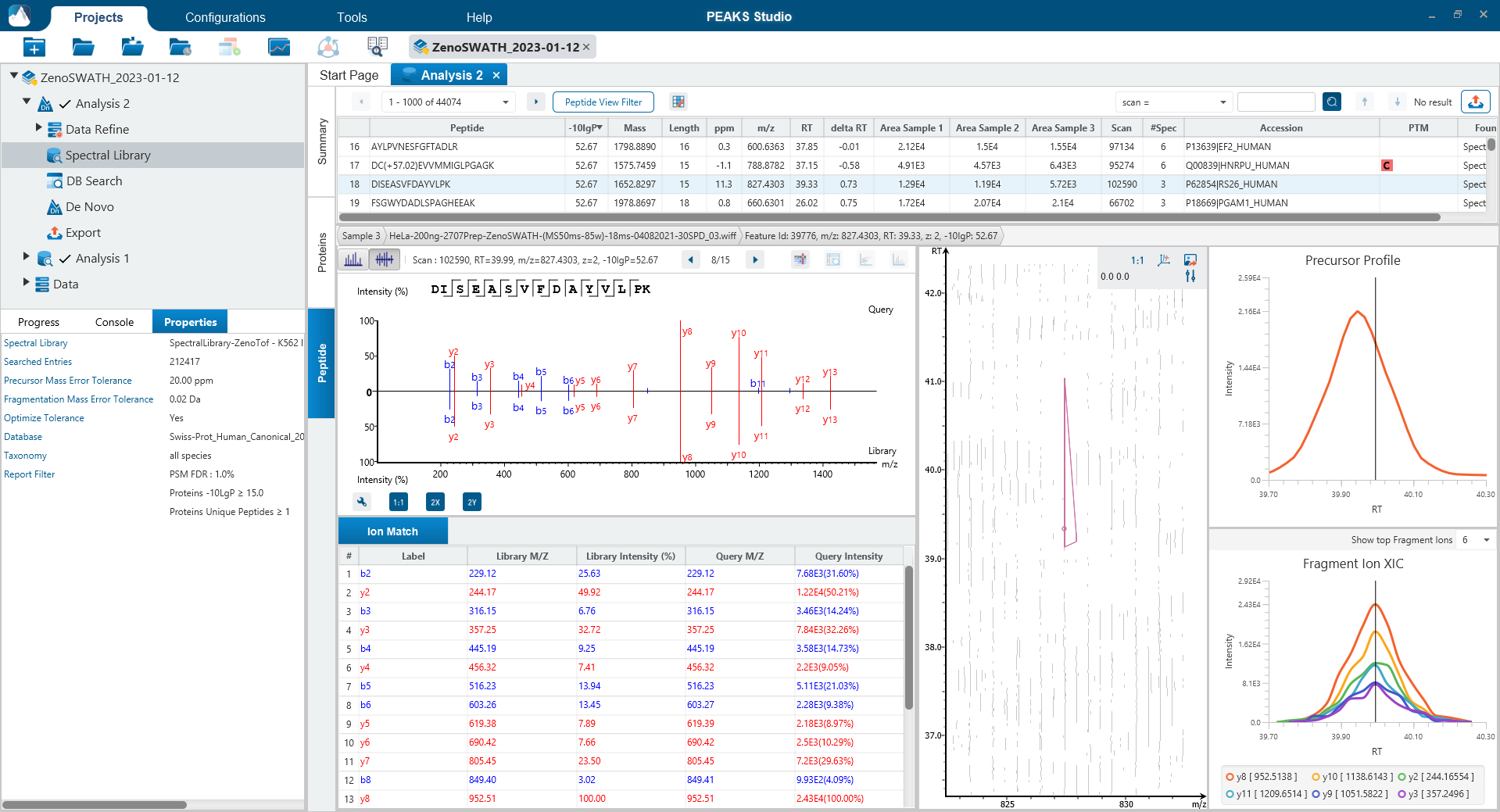
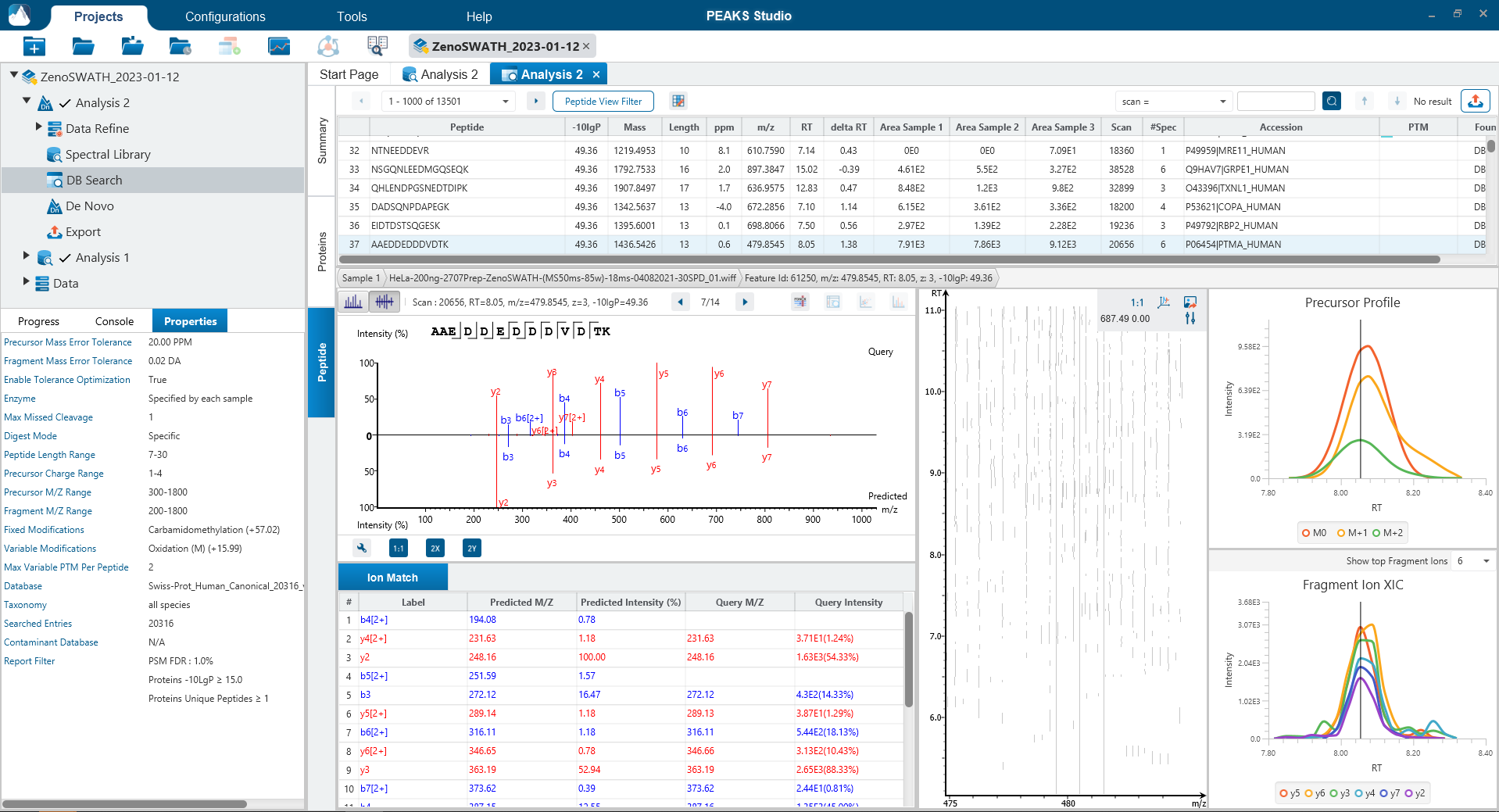
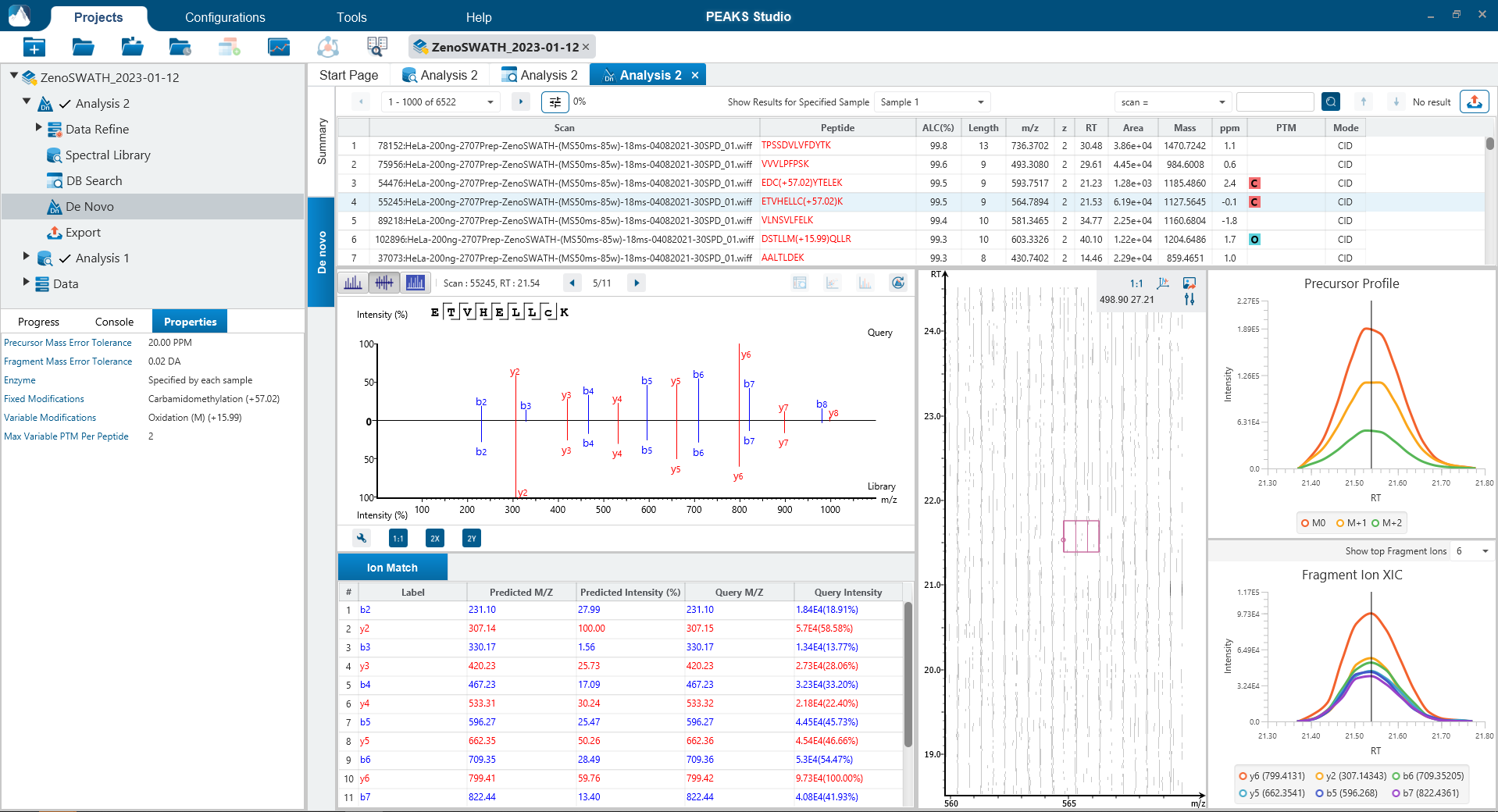
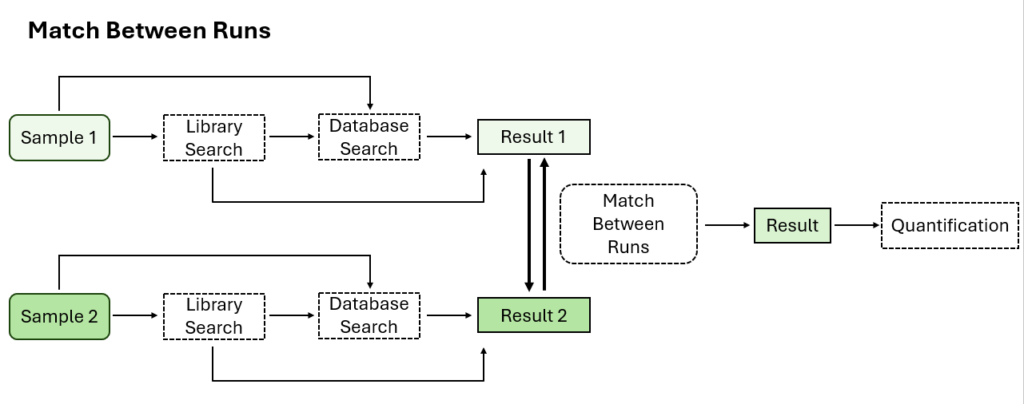
Optional Match Between Runs Step
Match between runs allows for alignment of features across multiple samples based on charge state, m/z, retention time, and ion mobility. This allows for transfer of MS2-idenifided peptides from one sample to another in the case where one is missing an MS2 identification to increase coverage.
In previous versions of PEAKS, match between runs was performed at the quantification step. Now in PEAKS 12, users have the option to perform match between runs at the identification step in the DIA workflow.
All-inclusive PTM support in DIA workflow
Identifying PTMs in a direct database search with DIA data presents many challenges due to the large search space and complex DIA spectra. By re-training a deep learning model and using a universal code that considers chemical formulas of PTMs, PEAKS 12 DIA workflow now supports the identification of any PTMs specified by the user. This will enable an increase in identification of modified peptides without requiring their entries in a spectral library.
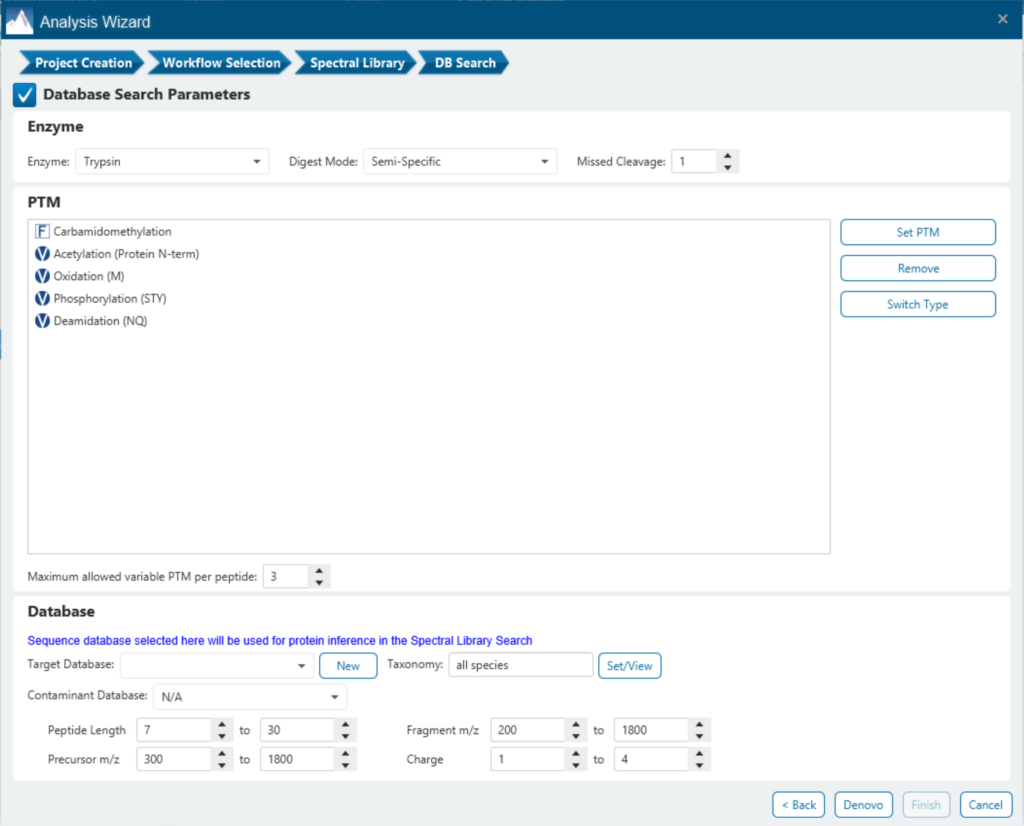
Add the PEAKS Q module for accurate and reproducibility quantification
DIA mass spectrometry produces highly reproducible quantification results compared to DDA methods. Spectral library, and direct database search results can be carried forward to a label-free quantification method. Fragment ions that follow the same elution profile as the precursor ion are included in the peptide abundance calculation for improved accuracy. The result is a robust set of quantification results with minimal missing values to help you discover biologically significant changes between conditions.
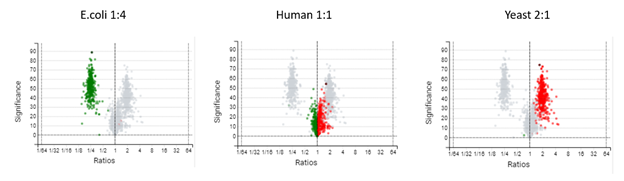
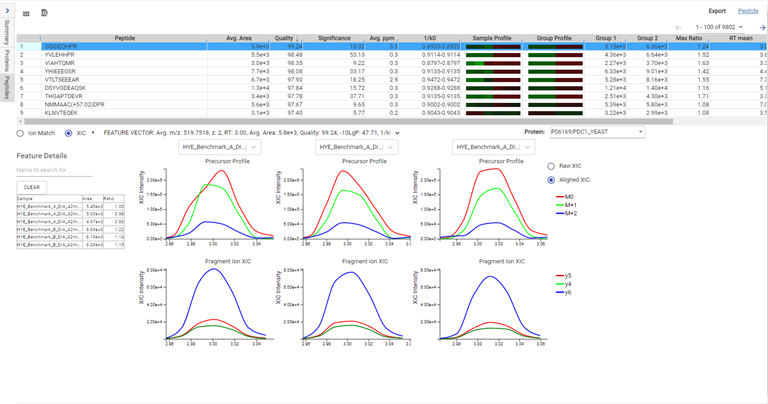
Label-Free Quantification
In the DIA LFQ workflow, users have the choice between two quantification modes, prioritizing either high precision (higher sensitivity) or high accuracy.


References & Resources
References
- Ludwig, C., Gillet, L., Rosenberger, G., Amon, S., Collins, B. C., and Aebersold, R. (2018). Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 14, 8126 (2018). https://doi.org/10.15252/msb.20178126
- Meier F, Brunner A.D, Frank M, Ha A, Bludau I, Voytik E, Kaspar-Schoenefeld S, Lubeck M, Raether O, Bache N, Aebersold R,. Collins B.C., Hannes L. Röst & Mann M. diaPASEF: parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods, 17, 1229-1236 (2020) https://doi.org/10.1038/s41592-020-00998-0
- Collins, B. C., Hunter, C. L., Liu, Y., Schilling, B., Rosenberger, G., Bader, S. L., et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 8, 291 (2017). https://www.nature.com/articles/s41467-017-00249-5
- Fernández-Costa C, Martínez-Bartolomé S, McClatchy D.B., Saviola A.J.,Yu N.K, Yates J. Impact of the Identification Strategy on the Reproducibility of the DDA and DIA Results. J. Proteome Res. 19, 3153–3161 (2020). https://doi.org/10.1021/acs.jproteome.0c00153
- Xin, L. Qiao R, Chen X, Tran NH, Pan S, Robinoviz S, Bian H, He X, Morse B, Shan B, Li M. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat. Commun. 13, 3108 (2022). https://doi.org/10.1038/s41467-022-30867-7