PEAKS IMS Add-On Module
Ion-Mobility Spectrometry Mass Spectrometry (IMS-MS) provides a compelling analytical workflow for complex biological and chemical mixtures by adding an additional dimension of ion separation. This technology can be maximised using the PEAKS IMS add-on module to address the limited depth of analysis that is provided using DDA data and the difficulty of multiplexing MS/MS spectra using DIA data. Using PEAKS IMS, ion mobility data can be analysed using all workflows provided in PEAKS, including de novo sequencing, database and spectral library searching, and quantification analysis.
Highlights
- Interactive data visualization tools for IMS-MS data projected on ‘m/z to RT’ plane or ‘m/z to 1/k0’
- Analysis of 4-D detected features
- Resolves overlapping precursor scans and chimeric spectra
- Supports 4-D labelled and label-free protein quantification
- DIA PASEF
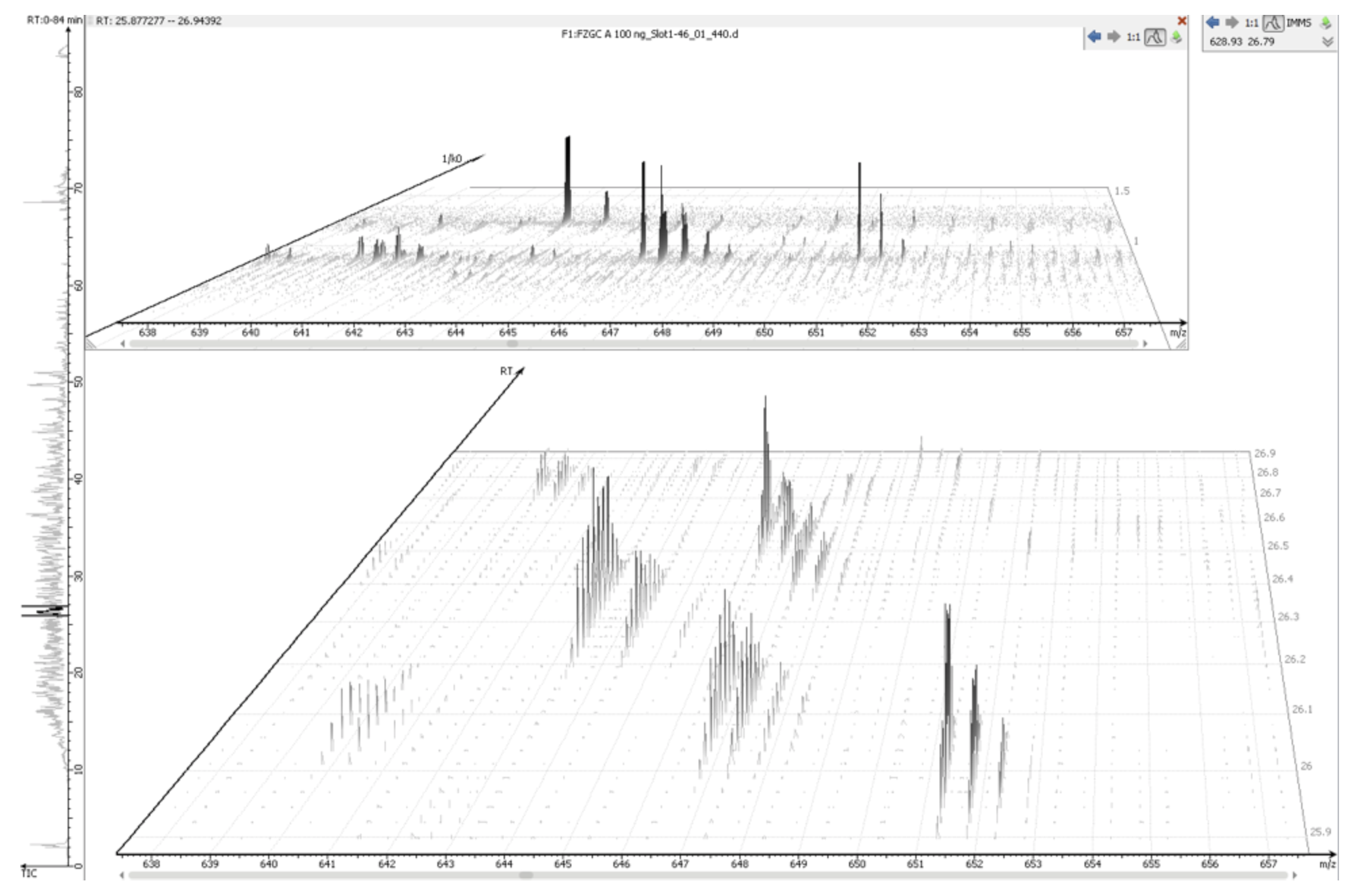
IMS-MS raw data can be loaded into PEAKS directly without the need for any preconversion steps. Once loaded into the software, the PEAKS IMS module allows the data to be analysed in four dimensions – m/z, retention time, intensity, and ion mobility. The combination of these vectors enables the data projection onto m/z-rt and m/z-1/k0 dimensions.
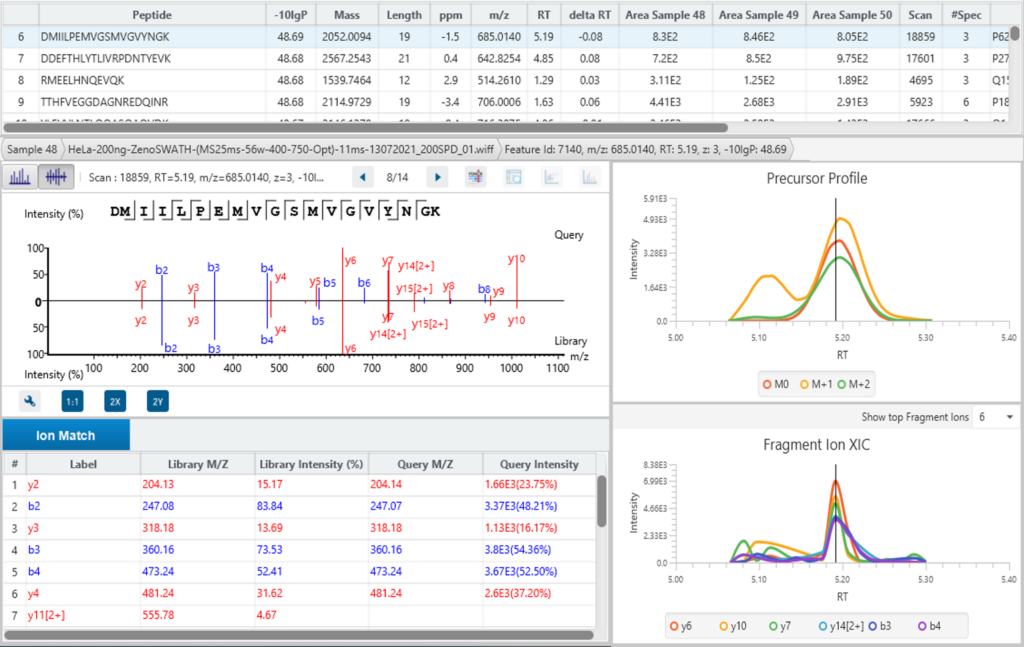
Using PEAKS IMS and the 4D information extracted from IMS-MS scans, PEAKS can accurately identify and quantify peptides, and subsequently proteins, from complex biological samples with a high degree of accuracy and sensitivity.
With ion mobility separation, two different peptides can be detected regardless of having very close m/z and retention time. Overlapping precursors would otherwise be disregarded without the increased sensitivity. By utilising all dimensions provided by IMS technology, PEAKS IMS can resolve these pain points commonly seen in discovery proteomics studies, like phosphopeptide positional isomers, by using a feature-based approach and 4D deconvolution. The example below shows co-eluted isobaric HEK Phosphopeptides which could only be separated in the ion mobility dimension.
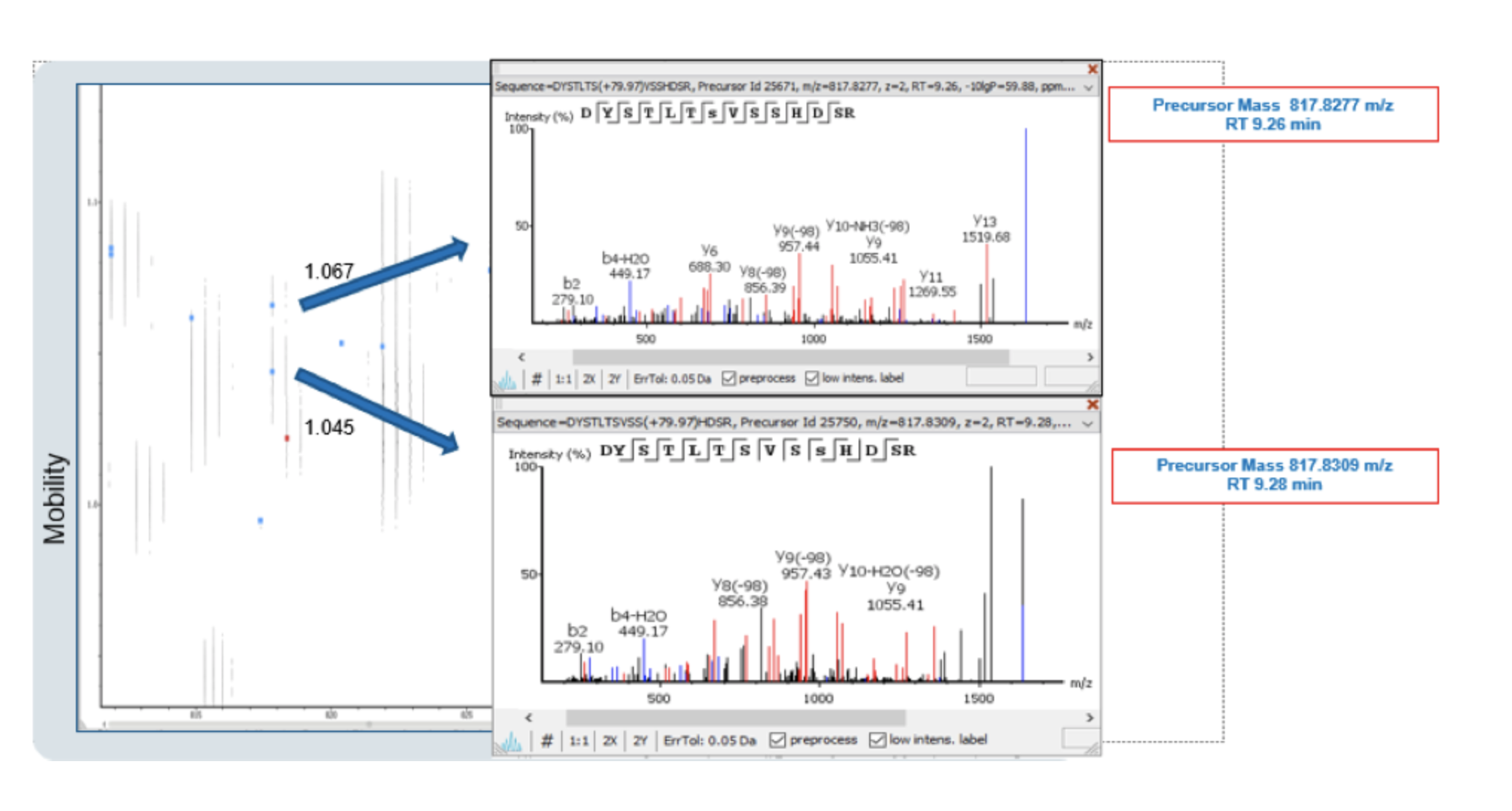
Without ion mobility separation, isobaric peptides are extremely difficult to identify and quantify by traditional proteomics workflows. By utilising IMS-MS data in combination with the PEAKS IMS add-on, the limitation to predict site localisation and quantification of the modification is overcome.
When paired with the PEAKS Q add-on module, PEAKS is able to utilise the 4D information from the PEAKS IMS module to quantitate the protein and peptide species using label-free or labelling techniques.
In addition to the retention time alignment performed in PEAKS Q, the PEAKS IMS module allows PEAKS Q to further perform feature alignment in the ion mobility dimension. According to Meier et al. (2018) in MCP, CCS values are highly reproducible and therefore alignment in this dimension ensures more accurate quantification results. PEAKS IMS uses a 4D feature alignment that maximises the dynamic range of identifiable and quantifiable peptides for analysis of biological samples.
Build spectral libraries using PEAKS IMS from PASEF data acquired in data-dependent mode and analyse diaPASEF (DIA) data acquired using Bruker’s timsTOF Pro.
Incorporating the ion mobility dimension into the data acquisition, timsTOF Pro allows for up to 100% sampling of the peptide precursor ion current using the new ion mobility-based Parallel Accumulation-Serial Fragmentation (PASEF) workflow. This is an ideal complement to data independent acquisition (DIA), a parallel data acquisition method that captures fragment ions over a large precursor mass range. Learn More
References
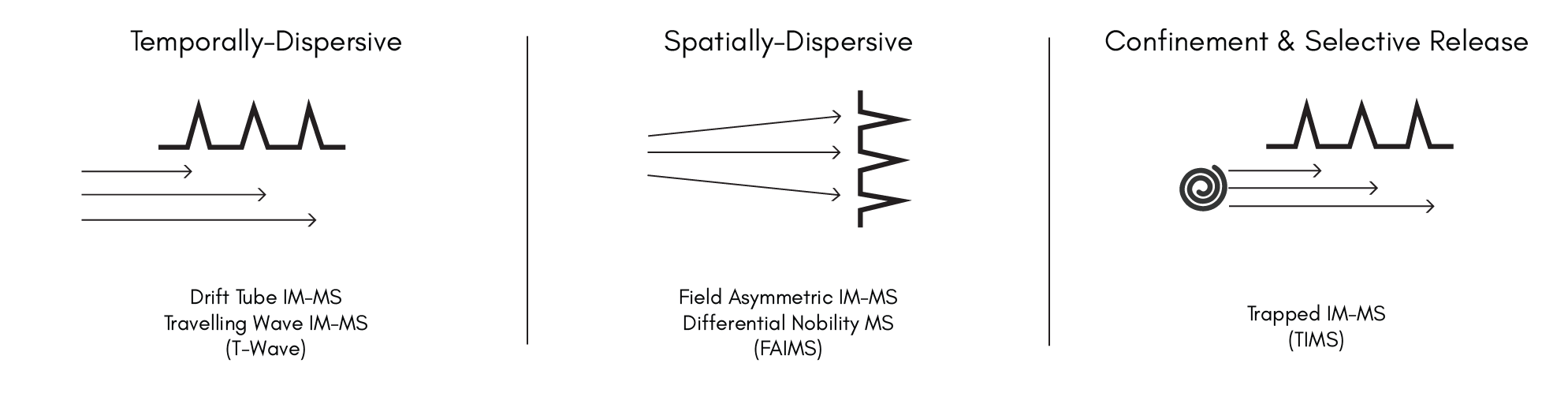
- May, J.C. and McLean, J.A. (2015). Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem., 87, 1422–1436 (2014). https://doi.org/10.1021/ac504720m
- Meier F, Brunner A.D., Koch S, Koch H, Lubeck M, Krause M, Goedecke N, Decker J, Kosinski T, Park M.A., Bache N, Hoerning O, Cox J, Räther O, Mann M., Online parallel accumulation – serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteomics, 17, 2534-2545 (2018). https://doi.org/10.1074/mcp.TIR118.000900