 de novo Sequencing Intro
de novo Sequencing Intro
Understanding the Fundamentals
Peptide de novo sequencing is the analytical process that derives a peptide’s amino acid sequence from its tandem mass spectrum (MS/MS) without the assistance of a sequence database. It is in contrast to another popular peptide identification approach – “database search”, which searches in a given database to find the target peptide. A clear advantage of de novo sequencing is that it works for both database and novel peptides.
Basic Principle
In a tandem mass spectrometer, the peptide is fragmented along the peptide backbone and the resulting fragment ions are measured to produce the MS/MS spectrum (Figure 1). Depending on the fragmentation methods used, different fragment ion types can be produced. The most widely used fragmentation methods today are Collision-Induced Dissociation (CID) and Electron-Transfer Dissociation (ETD). CID produces mostly b and y-ions; and ETD produces mostly c and z-ions.
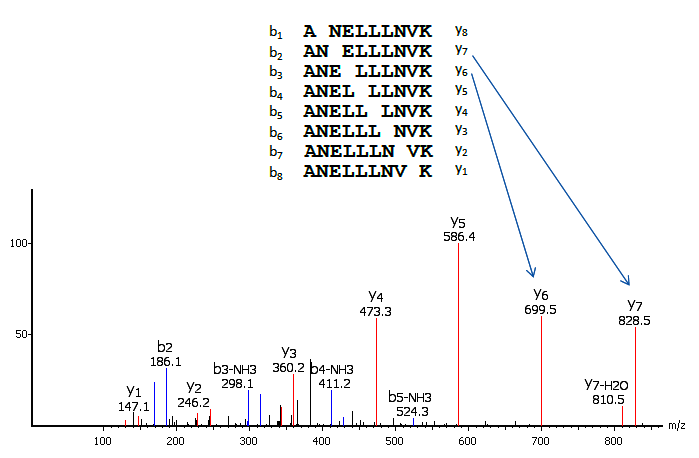
The main idea of de novo sequencing is to use the mass difference between two fragment ions to calculate the mass of an amino acid residue on the peptide backbone. The mass can usually uniquely determine the residue. For example, the mass difference between the y7 and y6 ions in Figure 1 is equal to 129, which is the mass of residue E. Similarly, the next adjacent residue between y6 and y5 can be determined as L by the mass difference. Such a process can be continued until all the residues are determined. A mass table of amino acids is provided for reference.
Thus, if one can identify either the y-ion or b-ion series in the spectrum, the peptide sequence can be determined. However, the spectrum obtained from the mass spectrometry instrument does not tell the ion types of the peaks, which require either a human expert or a computer algorithm to figure out during the process of de novo sequencing. During this process, a few factors can cause difficulties:
- Incorrect assignment of y and b ions.
- Some fragment ions are missing (such as b1 and y8 in Figure 1).
- Existence of other fragment ion types (such as the b3-NH3 ion in Figure 1).
- Existence of noise peaks in the spectrum.
- The same or similar mass of some residues may cause ambiguity (I=L and K=Q).
- The PTM (post-translational modifications) on the residues may contribute to the mass ambiguity, as well as complicate the peptide fragmentation pattern.
These factors can cause de novo sequencing to figure out only a partially correct sequence tag from the spectrum.
Auto de novo Sequencing
Manual de novo sequencing requires human experts and is very time consuming. A reliable auto de novo sequencing solution saves precious human time and greatly reduces the labour cost in labs. Automated de novo sequencing has been extensively studied in the bioinformatics community and multiple algorithms have been developed. Although the basic principle used by computer algorithms is the same as manual de novo sequencing, computer algorithms usually carry out the computation in a very different procedure than manual analysis.
First released in 2002, PEAKS Studio software has become the industrial standard software for automated de novo sequencing, and is well known for its accuracy, speed, and ease of use. The following video highlights the de novo sequencing features of PEAKS Studio.
With a Database
De novo sequencing was historically thought to be slow. Therefore it has been mostly used when the protein database was unavailable. However, with recent development in computer algorithms such as PEAKS, speed is no longer an issue. This makes de novo sequencing a viable choice for every mass spectrometry analysis in proteomics. Even when a database is available, de novo sequencing can contribute to peptide identification in the following ways.
- The matching or similarity between the de novo sequencing peptide and the database search peptide is a good indication that the database search result is correct. Therefore, de novo sequencing can be used to improve database search performance.
- The de novo sequencing peptides without significant database hits are possibly novel peptides in the sample, and deserve further examination, such as the finding of unexpected PTM or peptide mutations.
These considerations have been employed in the standard workflow of PEAKS Studio as illustrated in Figure 2.

References
- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M, Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry, Nat. Methods, 16, 63–66 (2019). https://doi.org/10.1038/s41592-018-0260-3
- Hughes, C., Ma, B., Lajoie, G.A. De novo Sequencing Methods in Proteomics. Methods Mol. Biol. 604 (2009). https://doi.org/10.1007/978-1-60761-444-9_8
- Ma, B. & Johnson, R. De novo Sequencing and Homology Searching. Mol. Cell. Proteomics. 11 O111.01490 (2012). https://doi.org/10.1074/mcp.O111.014902