Video Tutorials
![]()





Videos in this section introduce basic functions of PEAKS Studio and help you better understand and use the software for mass spectrometry data analysis.









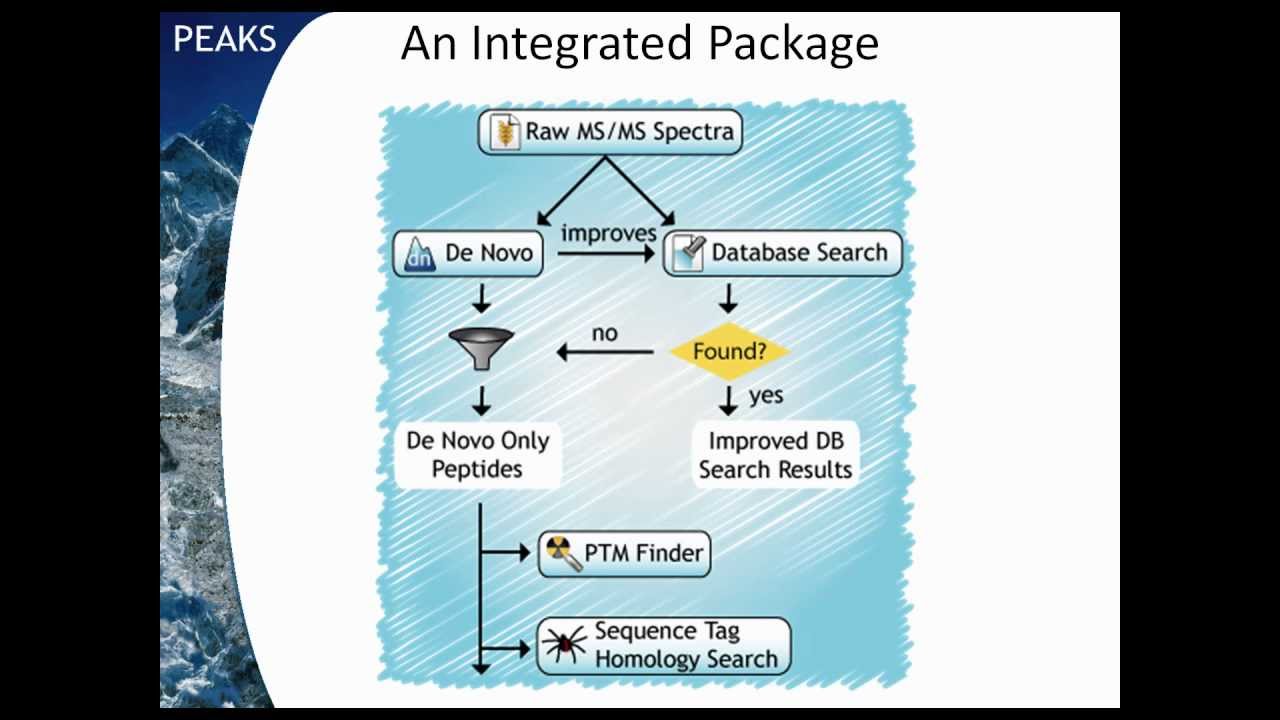

Find a few past webinar recordings here! We hold free webinars regularly to show what PEAKS Studio can do for your research and how to use this tool.