
Complete Solution for Bottom-Up Proteomics
PEAKS® Studio 13 offers a complete bottom-up proteomics solution with increased accuracy, sensitivity, and speed. Updated workflows for a variety of applications, such as in depth canonical and non-canonical peptide and protein identifications, make PEAKS® a unique solution. From DDA to DIA data support, PEAKS® Studio 13 provides a comprehensive solution to bring your research to new heights!
One-Stop
Discovery and Targeted Proteomics
Comprehensive
and Confident
DDA Solutions

High Accuracy and Sensitivity DIA Solutions
High Precision
PRM Solutions
de novo and
DeepNovo
Algorithms
Vendor Neutral
Software
Contact Us to Add PEAKS Studio 13 to your lab!

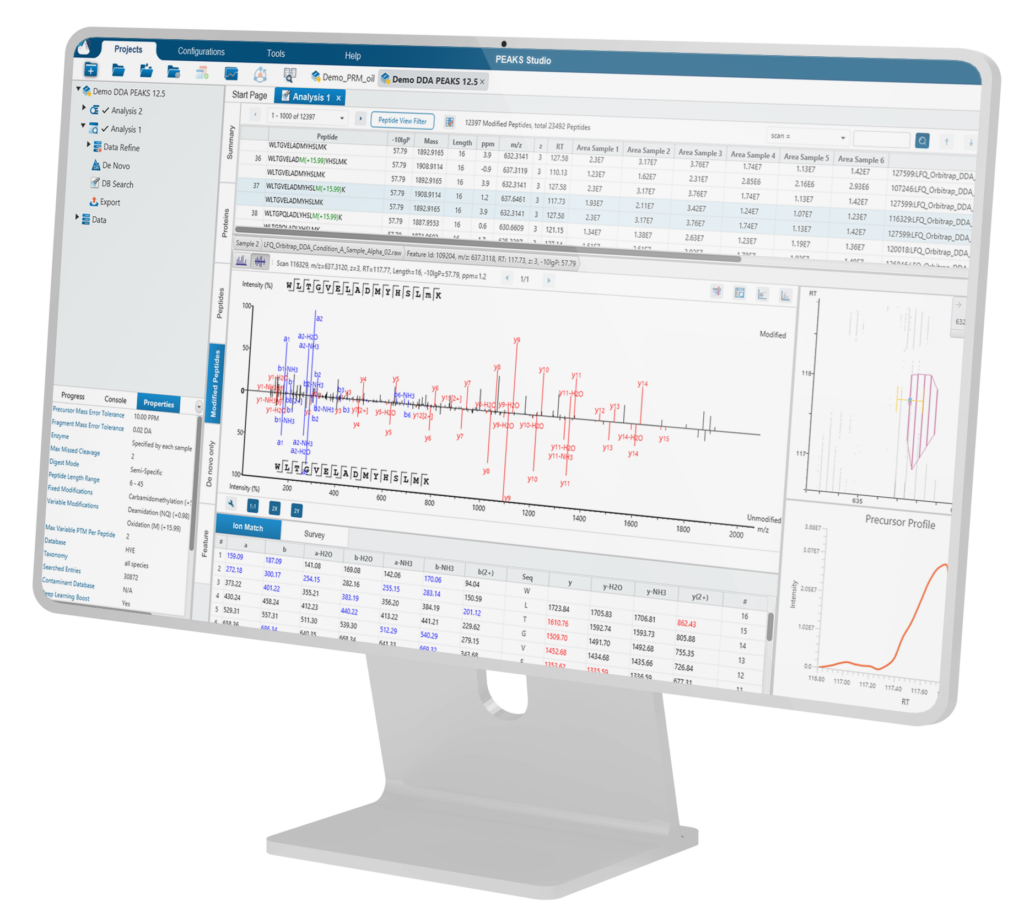
DDA and DIA Done Right: Multi-Engine Powerhouse
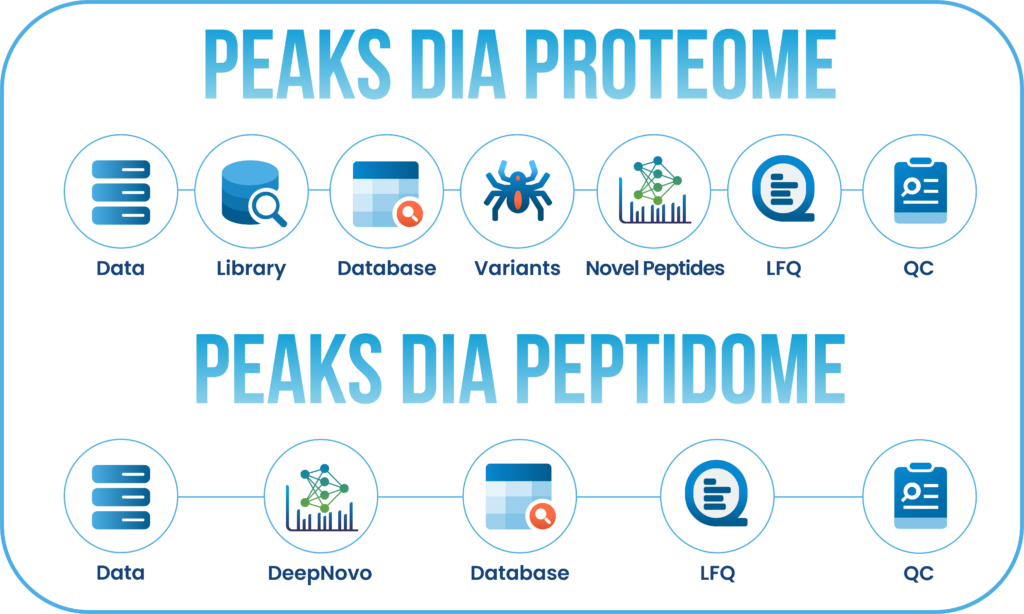
In PEAKS 13, the DDA and DIA workflows have been streamlined into two powerful options: Proteome and Peptidome.
The Proteome workflows support traditional protein identification and quantification, with results focused at the protein level. Meanwhile, the Peptidome workflows leverage DeepNovo’s deep learning-based de novo sequencing, combined with peptide database searches and sequence variant analysis, to deliver insights at the peptide level.
Users can customise their analysis workflows, selecting which steps to run and adding or modifying steps at any point. Results are organised into intuitive result nodes, making it easier to interpret, explore, and validate your data.

Key Features
PEAKS 13 uses the latest PEAKS algorithm for all analyses, including data loading/refinement, identification and quantification. Deep learning-boosted identification workflows are available for both DDA and DIA analysis for increased identification rates of over 10%.
DIA Workflow

LC/MS Data
Refine View

Feature-Based Identification
DeepNovo
Peptidome
Confident
PTM/Mutations
Quality Control (QC) Function
DIA Feature
View
Quantification
View

Hybrid DIA
and PRM
Protein &
Peptide View
Streamlined Workflow with Direct Database search for DIA
PEAKS Studio 13 offers a unique DIA workflow to maximise identification of peptides by integrating four methods: spectral library search, direct database search, de novo sequencing, and sequence variant analysis with up to 20% increase in identification and quantification, and faster processing times for CPU and GPU resources.
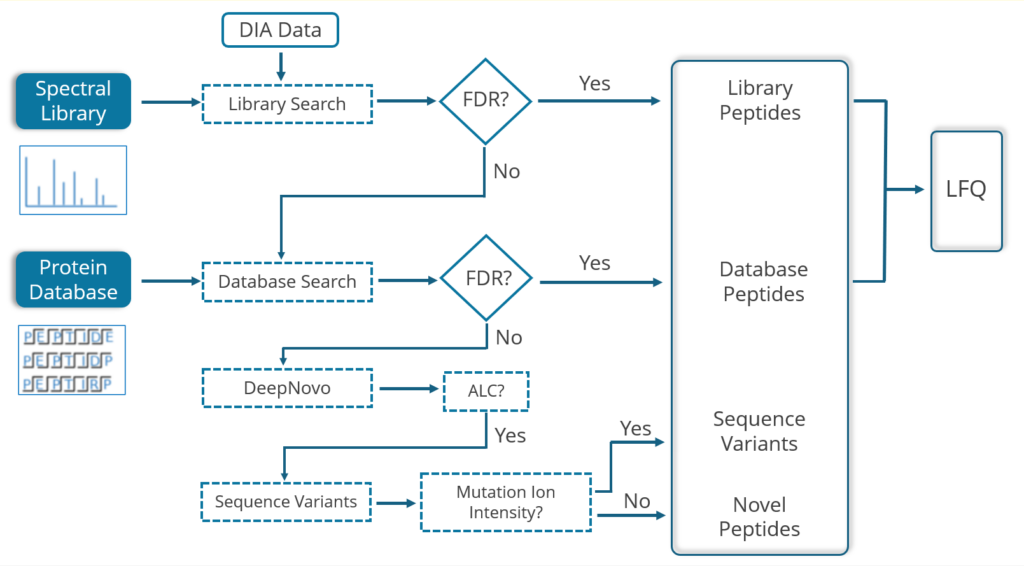
- A library search is performed against a predefined spectral library. Peptide spectra without a library match can be directly searched against a protein database.
- A protein sequence database is directly searched with DIA data. Advanced machine learning algorithms allow improved accuracy and sensitivity of peptide identification. During this step of the pipeline, PEAKS 13 DIA workflow now supports the identification of any PTMs specified by the user. This will enable an increase in identification of modified peptides without requiring their entries in a spectral library.
- Unmatched spectra from the database search are de novo sequenced.
- High-scoring de novo peptides are mapped against the database to find sequence variants using the SPIDER algorithm.
- Identified peptides from both the spectrum library search and protein sequence database search can be used in a quantification analysis.
The library search and the database search methods are optional. Users can conduct only a library search, a library-free direct database search, or both steps in sequence.
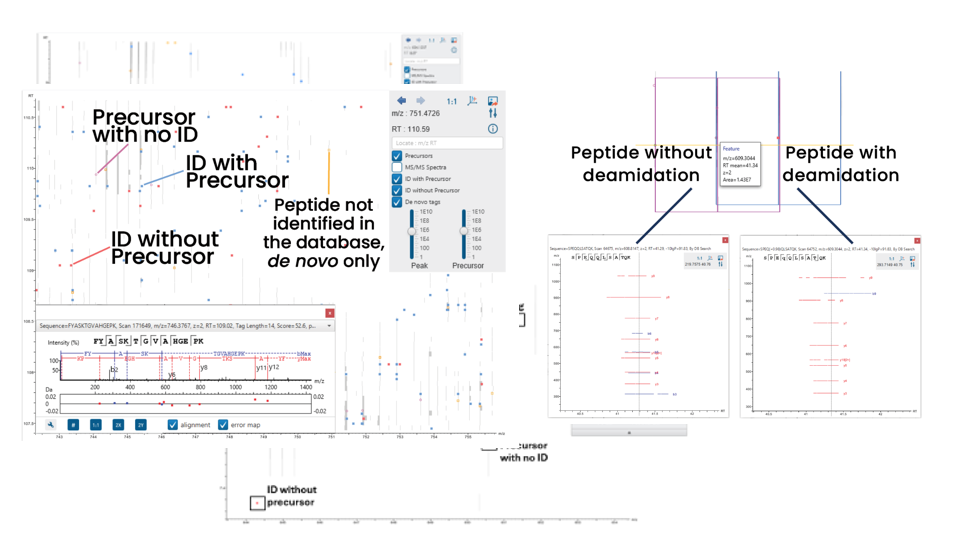
LC/MS Data Refine View
Users can now easily visualise precursors, identifications with and without associated precursors, and de novo tags on the LC/MS map for improved result validation.
Additionally, the peptides tab and LC/MS map are more seamlessly integrated. Click on one or more identifications to show and compare the supporting fragment ions. This makes it easy to verify overlapping peptide identifications.
The supporting fragment ions are also shown in an LC/MS snapshot in the peptides tab. Users can also click to show the identification on the full LC/MS map.
Feature-Based Identification
- MS1 Feature-based Identification increases sensitivity and peptide ID efficiency.
- Designed for DDA technology to improve reproducibility.
- Integrate database search and de novo sequencing to extend in-depth analysis.
- Activate Deep learning-boost in PEAKS DDA workflows to maximise peptide ID efficiency.
Learn more about the advantages de novo sequencing brings to your research.
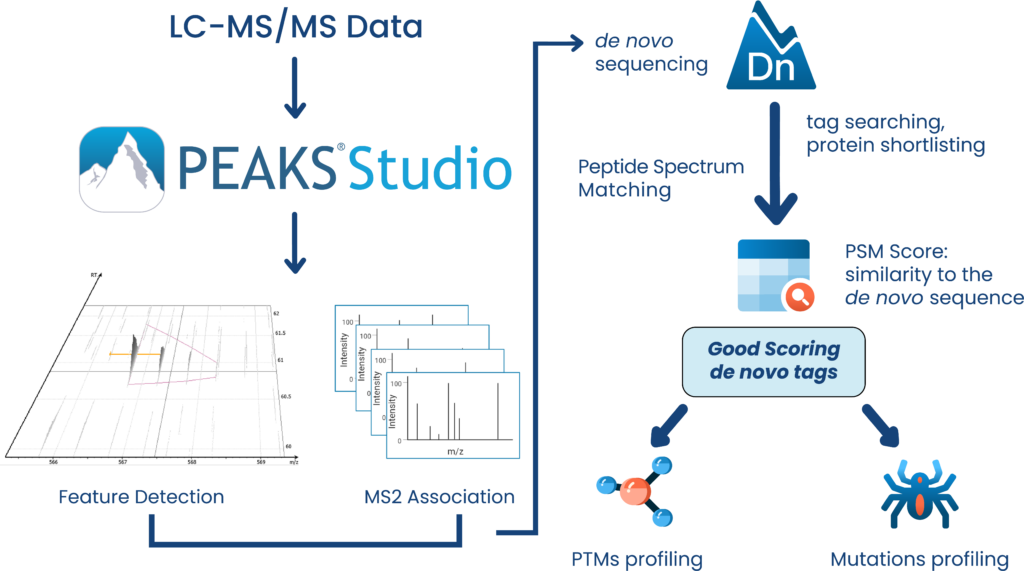
PEAKS DeepNovo Peptidome: Advanced solution Immunopeptidomics
This newly developed solution is a specialised workflow for peptidomics data that combines database searching, de novo sequencing, and identification of mutated peptides. Learn more
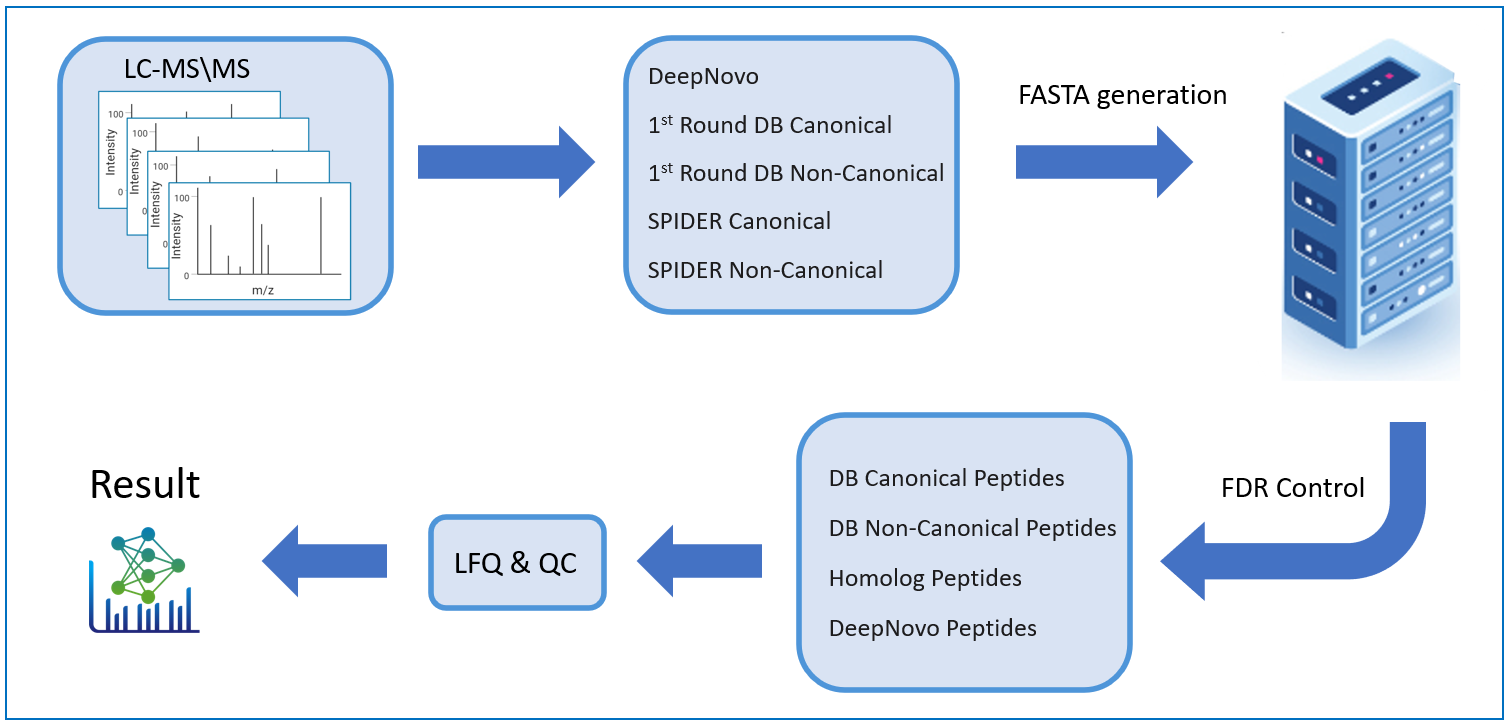
Next level confidence: confident PTM/mutation sites identification based on amino acid level evidence
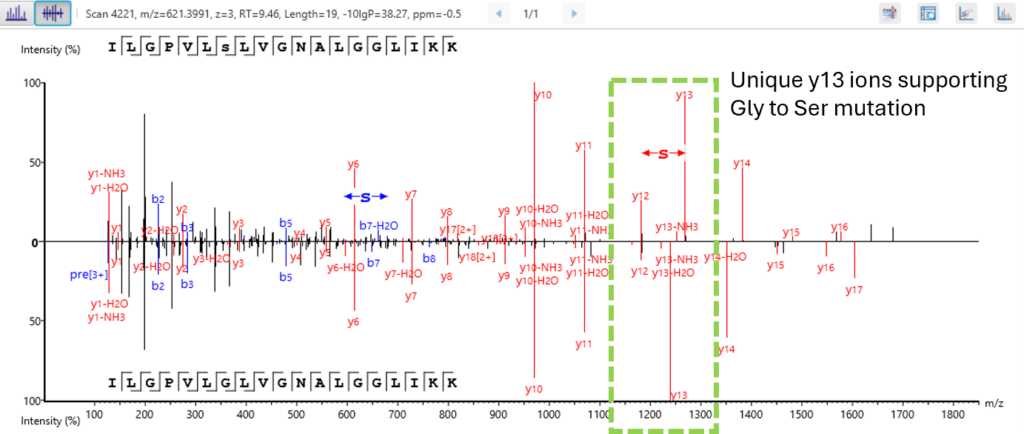
The Post-Translational Modification (PTM) and Mutations Analysis workflow offers next-level confidence by enabling the identification of PTM and mutation sites with high precision, grounded on amino acid level evidence.
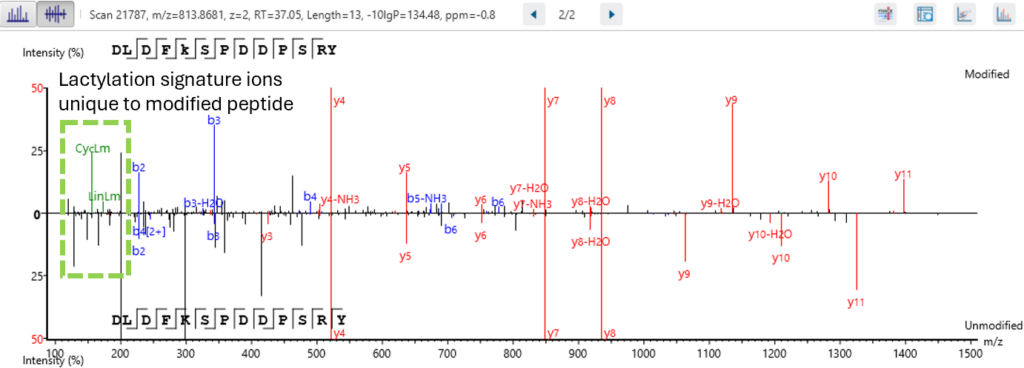
This advanced capability allows researchers to pinpoint specific modifications and mutations within proteins, providing a detailed and accurate understanding of protein function and regulation. By leveraging robust algorithms and comprehensive data analysis, this workflow ensures that PTM and mutation sites are identified with exceptional confidence, facilitating deeper insights into protein dynamics and disease mechanisms. This enhanced analytical power is crucial for advancing our understanding of biological processes and developing targeted therapeutic strategies.
Quality Control (QC) Function for in-depth analysis from raw data to results
With the Quality Control (QC) analysis in PEAKS® Studio 13 users can assess statistical information of the raw data and/or results and gain beneficial insight into the attributes of the LC-MS acquisition. This automated tool is designed for both, DDA and DIA data and will supply the elements to determine the quality of the data and evaluates the setup of the experiment.
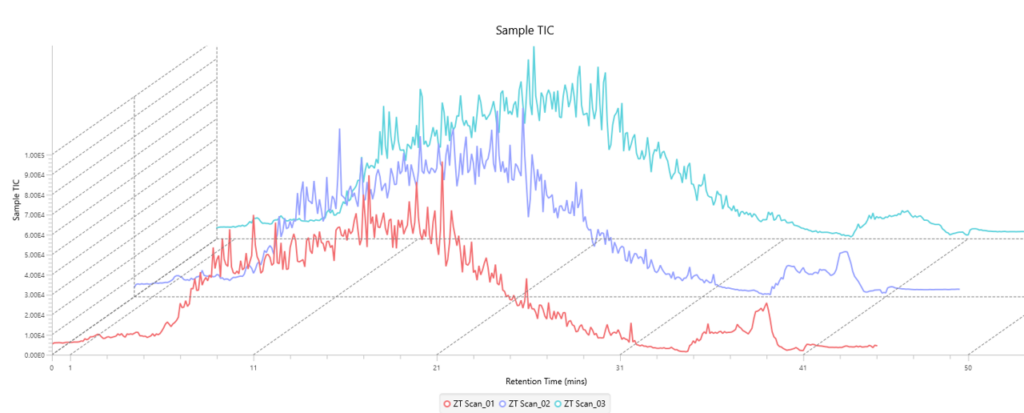
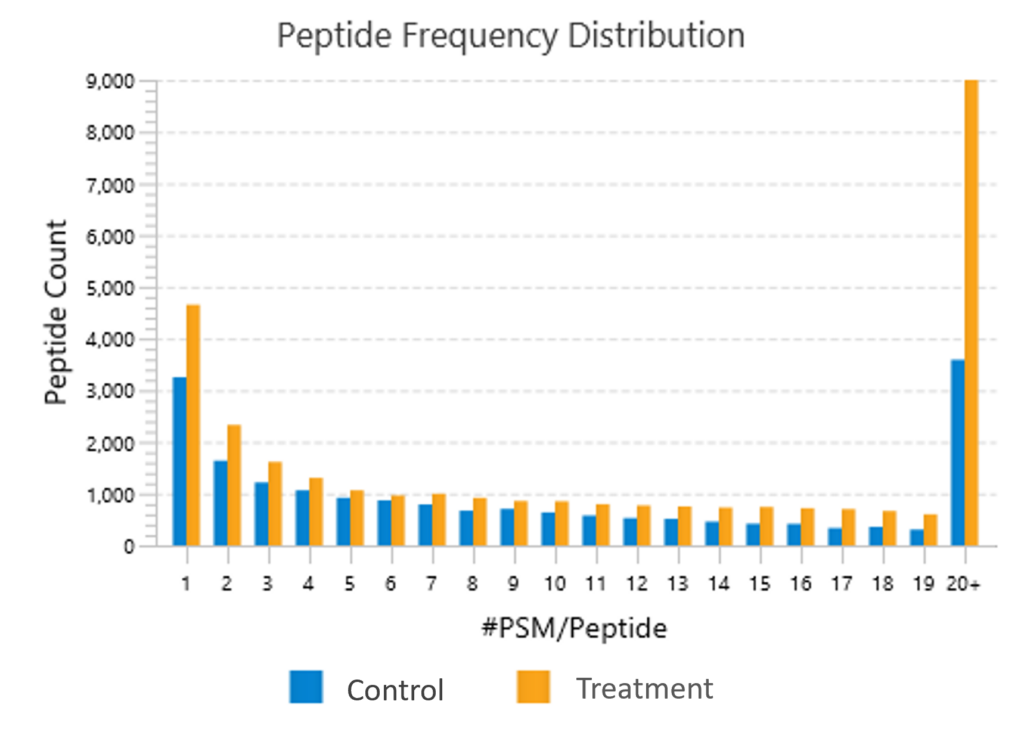
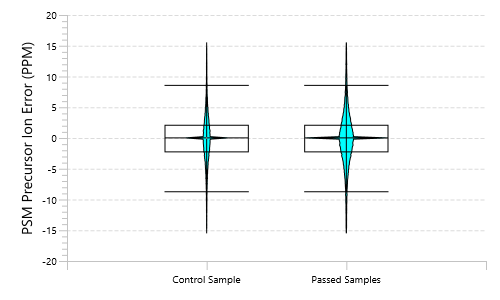
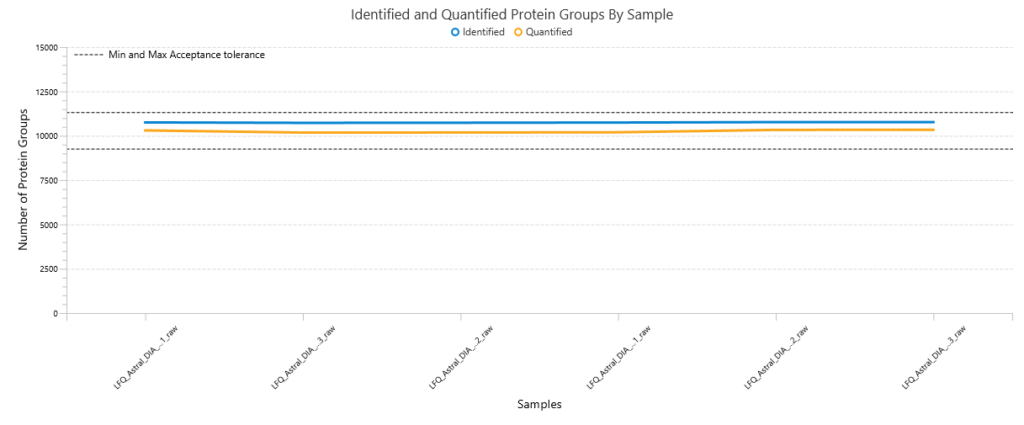
The DIA Feature View
DIA Feature View stems from PEAKS dedication to provide a full result transparency allowing the user to investigate, validate, and confirm the precursor. All features, with ID and without ID, are displayed with all fragment assignments, mirror plots to show correlation of empirical and predicted fragments, MS1 and MS2 feature views.
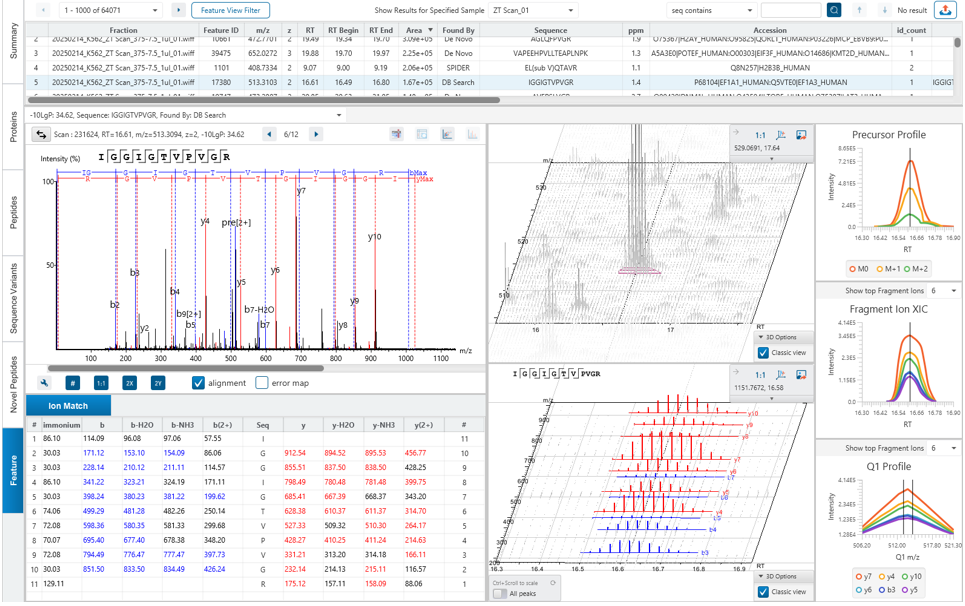
The Quantification View
With the add-on module of PEAKS Q, PEAKS Studio also determines relative protein abundance changes across a set of samples simultaneously and without the requirement for prior knowledge of the proteins involved. In PEAKS 13, peptides found through PEAKS PTM or SPIDER searches will be included in the quantification step.
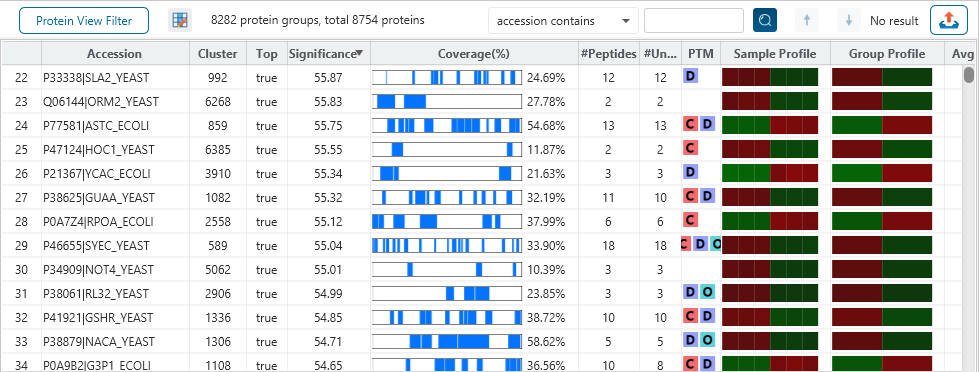
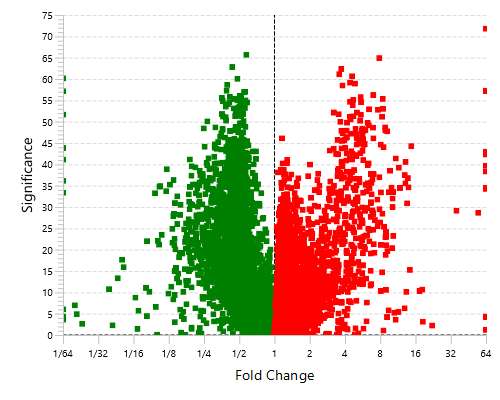
Highly differentially expressed proteins between two groups are identified by statistical analysis tool (fold change >2, FDR <0.01) and displayed in a heatmap format.
If “Feature-based” quantification is selected, then the LFQ result will contain a new “de novo only” tab with quantification results for peptides which were not matched to the database.
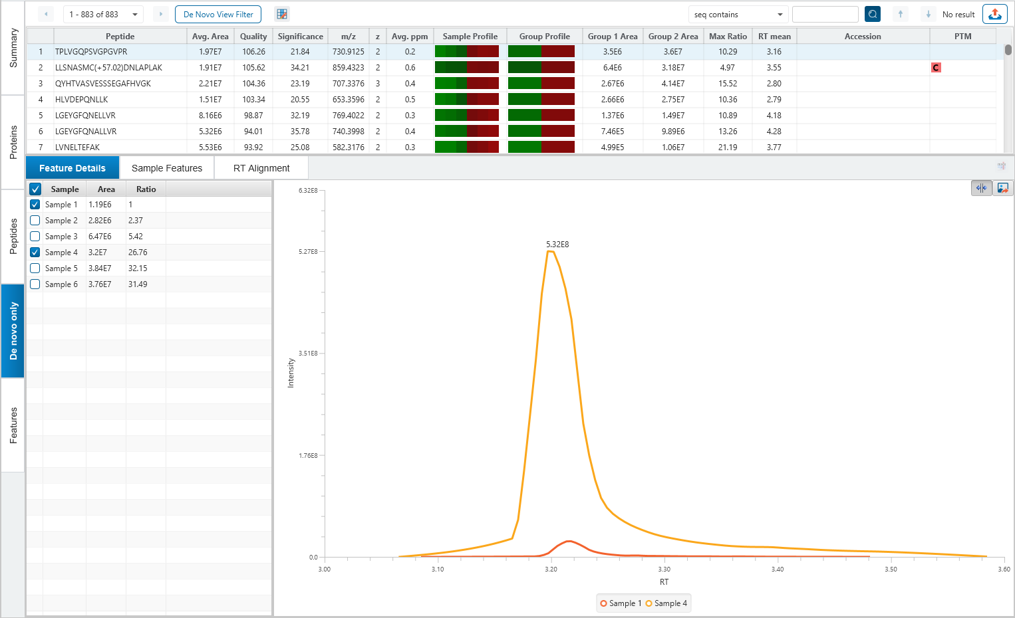
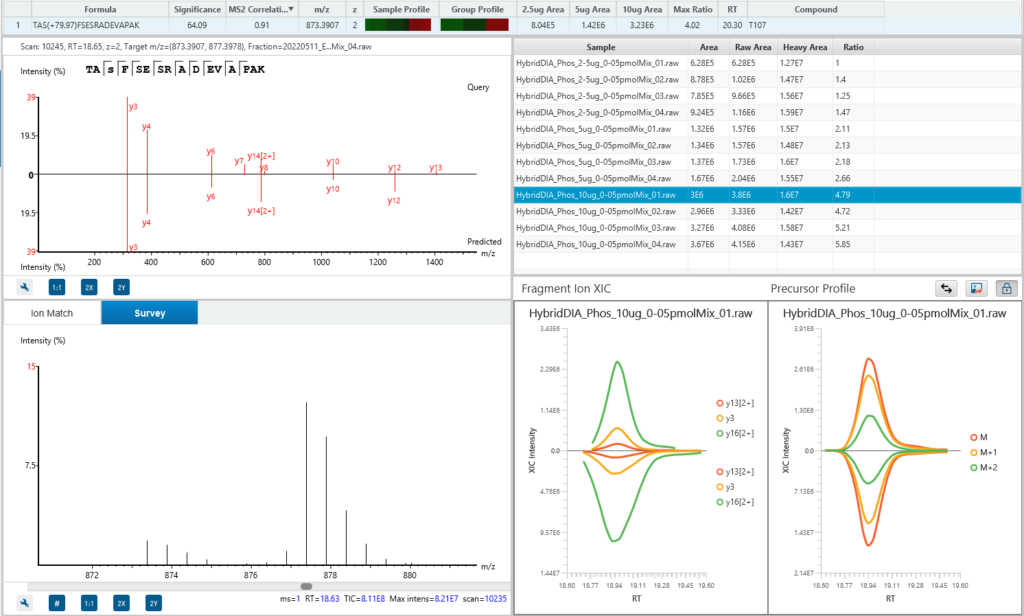
Hybrid DIA and PRM
The Hybrid-DIA technology by Thermo, available exclusively in PEAKS Studio, combines targeted and discovery-driven approaches, making it ideal for clinical proteomics applications.
Hybrid-PRM/DIA is a new intuitive data acquisition strategy that enables enhanced sensitivity for a specific set of analyses by the intelligent triggering of multiplexed parallel reaction monitoring (PRM) in combination with the discovery driven digitisation of the clinical biospecimen using DIA. Heavy labelled reference peptides are utilised as triggers for PRM and monitoring of endogenous peptides.
PEAKS PRM
PEAKS 13 features a new PRM Targeted Quantification workflow that is precise and reliable for quantifying specific peptides in complex biological samples.
This targeted approach allows researchers to focus on predefined peptides of interest, ensuring accurate and reproducible quantification even in the presence of interfering substances. The new PRM Targeted Quantification capability significantly improves the workflow’s sensitivity and specificity, making it an indispensable tool for detailed proteomic studies and biomarker validation. By integrating this advanced feature, the PRM Workflow offers a robust solution for high-confidence absolute quantification in targeted proteomics.
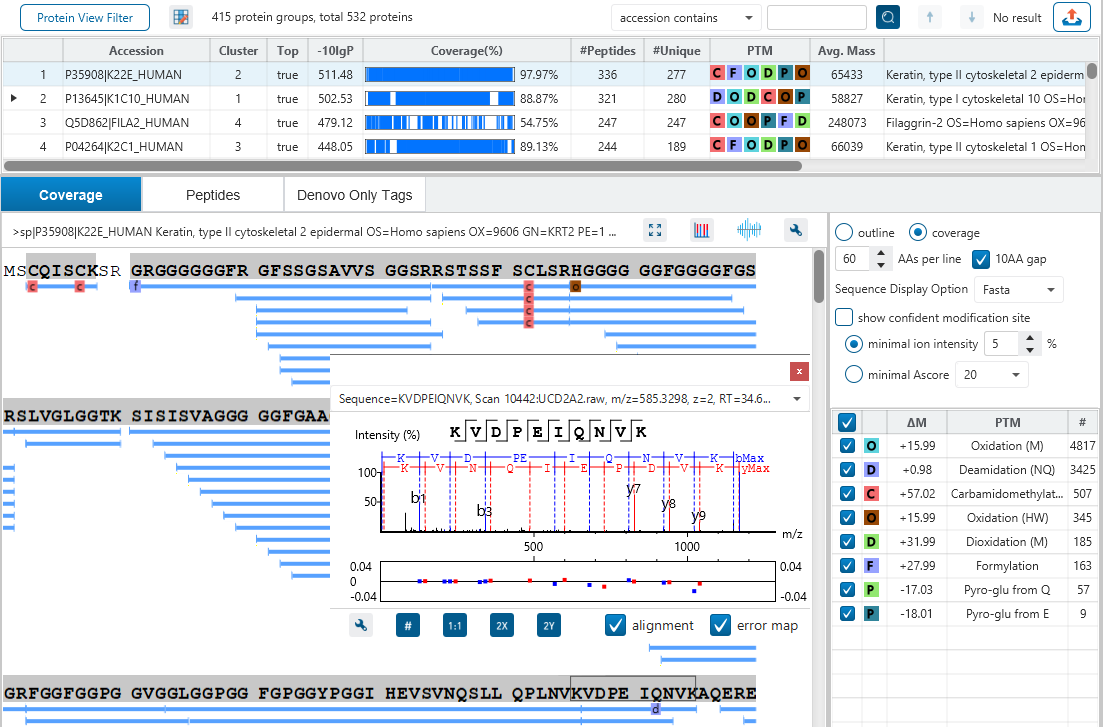
The Protein View
The Protein View presents protein profiling across complex biological samples in both DDA and DIA workflows. For each protein, the Sequence Coverage View displays a peptide map with spectrum annotation for validation. With PEAKS traditional de novo-assisted database search, users easily view identified peptide sequence in blue, while a grey bar indicates a de novo only tag match.
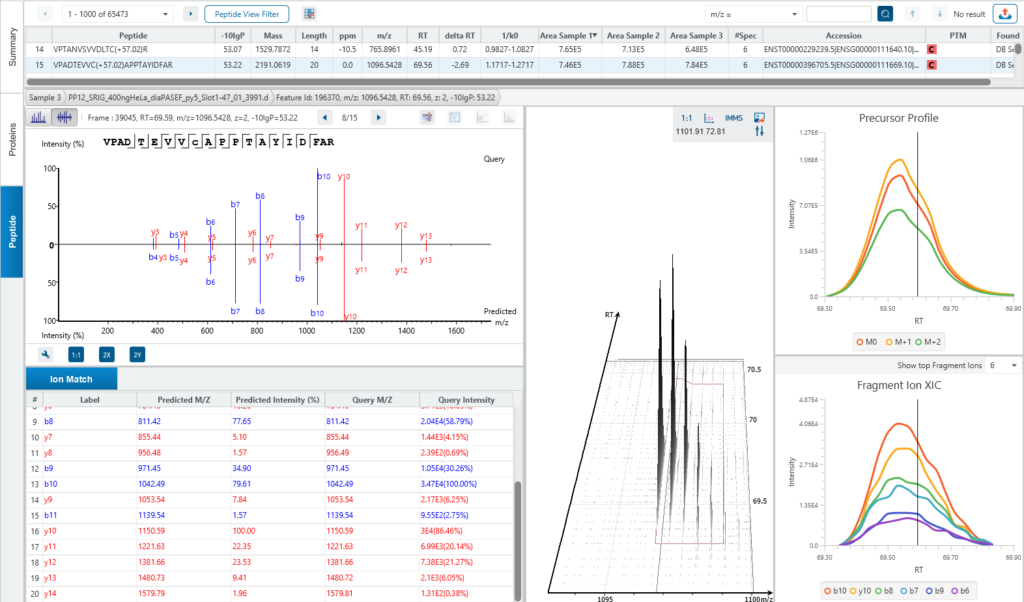
The Peptide View
The Peptide View provides a list of identified peptides with the abundance from MS1. For each modified peptide, the confidence of modification site (Ascore) is associated.
Unmatched Customer Service
✅ Fast, expert responses to your PEAKS questions
Our in-house scientists respond quickly to ensure you never hit a roadblock in your workflow or research timeline.
✅ Frequently updated tutorial resources
Access to step-by-step guides, walkthroughs, and webinars to get the most out of PEAKS Studio for you and your team.
✅ Continuous user-feedback-driven updates
We listen. Your insights and queries directly shape our future releases and platform improvements.

Learn More About
Licence Information
The PEAKS Studio licence can be scaled to address your lab’s requirements.
- Desktop – 24 threads, capable of processing across up to 24 cores
- Workstation – 48 threads, up to and across 48 cores
Configuration
PEAKS Studio 13 is recommended to be installed on 64-bit Windows operating systems with Windows 10 or later.
Recommended Configurations:
Desktop License: 30+ threads processors and 64 GB+ of RAM with compatible GPU (described below)
Workstation License: 60+ threads processors and 128 GB+ of RAM with compatible GPU (described below)
e.g. Intel Core i7/i9/Xeon or AMD Ryzen 7/9/Threadripper processors
For running DeepNovo, it is required that the machine is equipped with an NVIDIA GPU with CUDA compute capability ≥ 8 and at least 8GB of dedicated memory.
For running DIA Database Search, it is recommended that the machine is equipped with an NVIDIA GPU with CUDA compute capability ≥ 5 and at least 8GB of dedicated memory.
The GPU must be updated to CUDA version 12.3 or later.
References & Resources
References
- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M, Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry, Nat. Methods, 16, 63–66 (2019). https://doi.org/10.1038/s41592-018-0260-3
- Tran NH, Zhang X, Xin L, Shan B, Li M. De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. U.S.A. 114, 8247-8252 (2017). https://doi.org/10.1073/pnas.1705691114