
PEAKS Studio 13 introduces redesigned DDA and DIA workflows for streamlined and user-friendly experience with enhanced algorithms. PEAKS DIA offers highly competitive processing times and improved performance for a deep data insight with novel peptides and sequence variants. A full result transparency is now available for DIA as well through a new Feature tab. Please check all new features below!
Unmatched DIA Performance
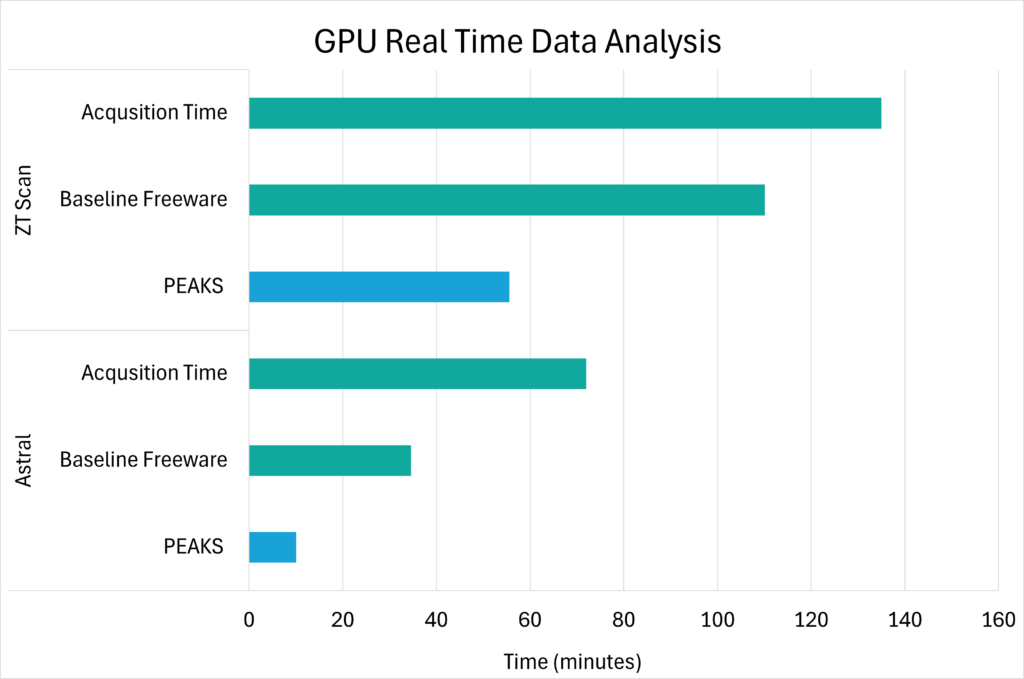
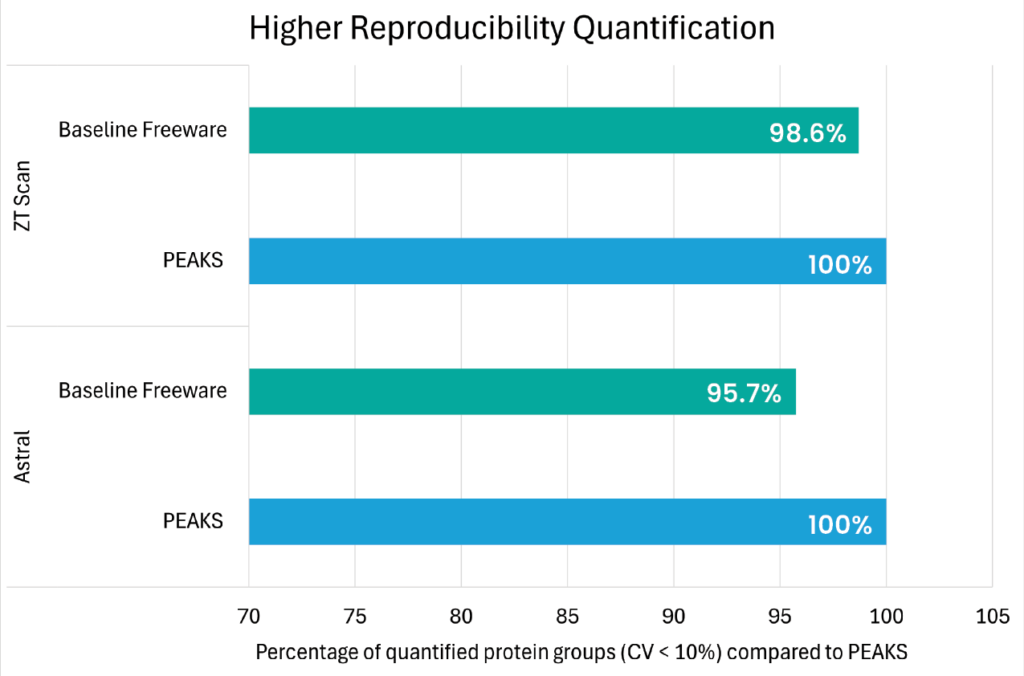
Our enhanced DIA solution will accelerate your discovery ensuring accurate and comprehensive result to provide complex biological insights!
Full Result Transparency
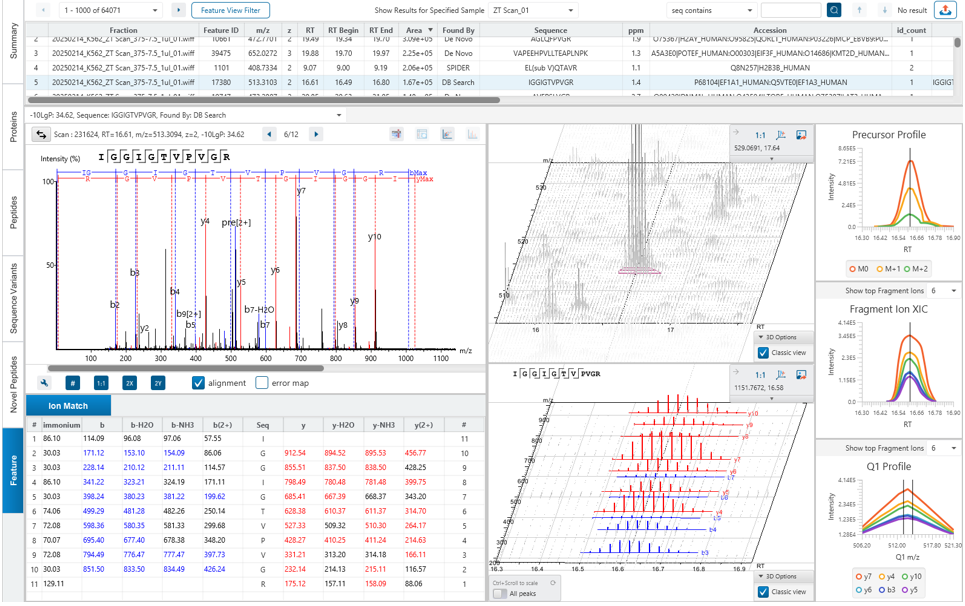
The New Feature tab gives an overview of all feature associations with each DIA identification step and allowing the user to investigate, validate and confirm the result.
Unlock Dark Proteome with Sequence Variants and Novel Peptides
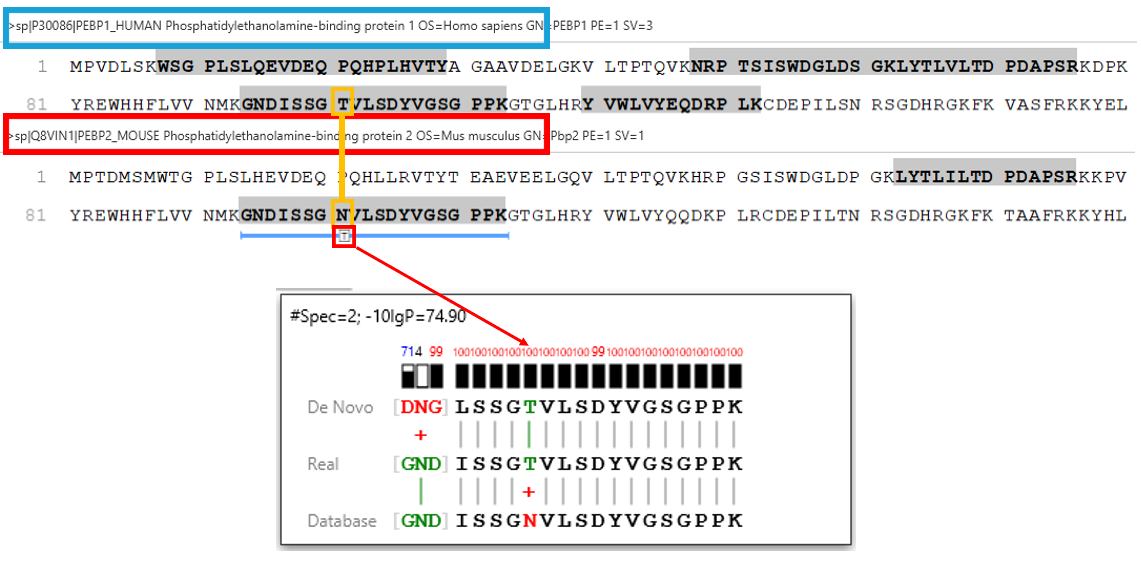
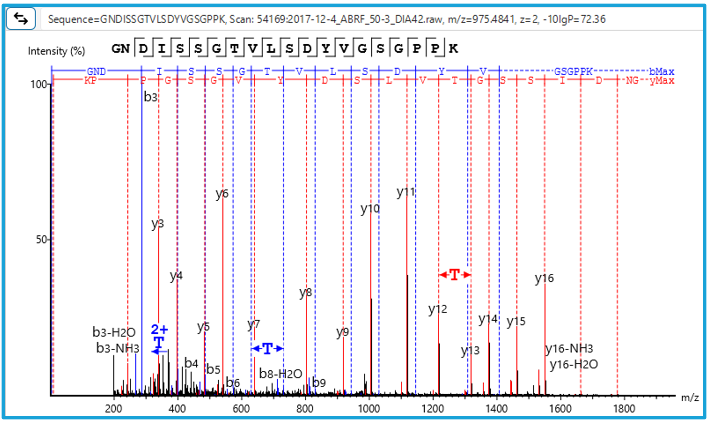
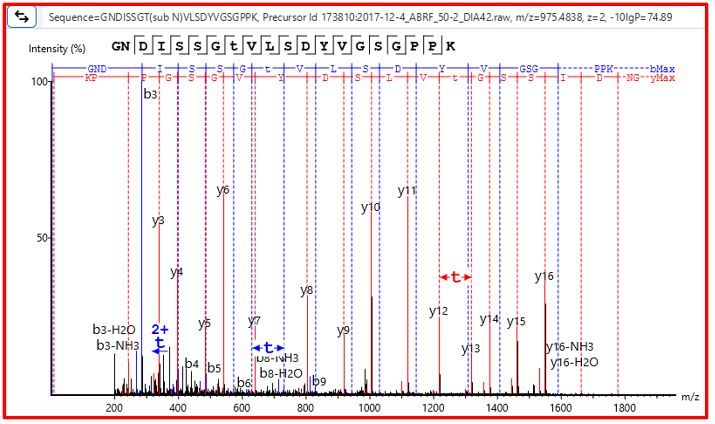
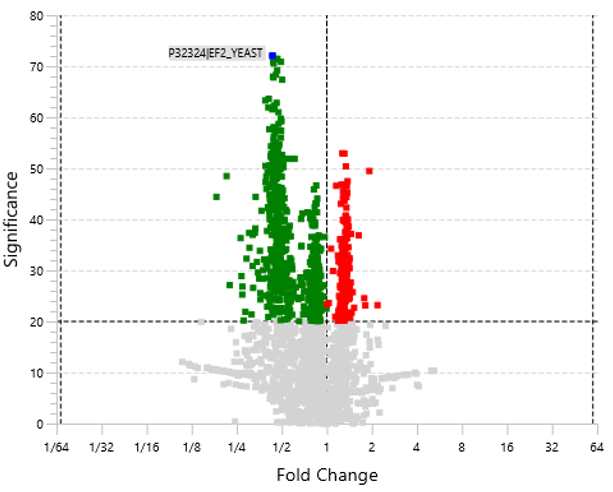
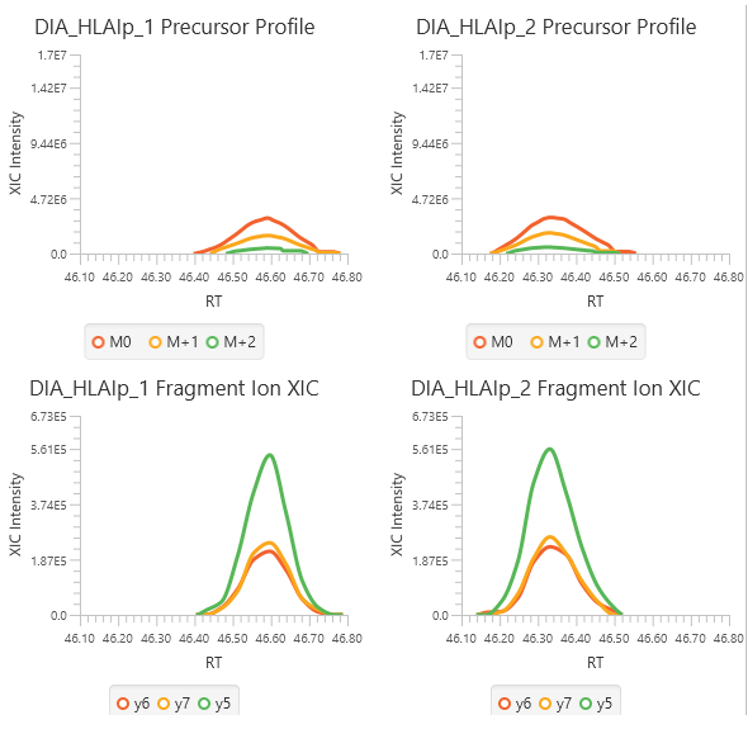
Human tryptic digest searched against Human and Mouse databases, Human peptide identified in the database search while the Mouse peptide is identified using a new DIA SPIDER as a mutation:
Other New Features
- Faster processing times for both CPU and GPU run analyses.
- Unprecedented performance, improved sensitivity and accuracy.
- Deeper discovery with Sequence Variants with Novel Peptides.
- Full result transparency via Feature tab.
- Enhanced protein and peptide scoring for higher confidence.
- Protein quantification using Top3 or MaxLFQ.
- Added optional Precursor FDR/score filter to apply additional result filtering.
- Use of the powerful GraphNovo deep learning sequencing.
- DIA Peptidome now offers insight into Sequence Variants.
- LFQ includes Database found peptides and Sequence Variants.
- Redesigned the DDA Proteome and Peptidome workflows in a streamlined workflow, improving usability and flexibility.
- Added quantification for PEAKS PTM and SPIDER result.
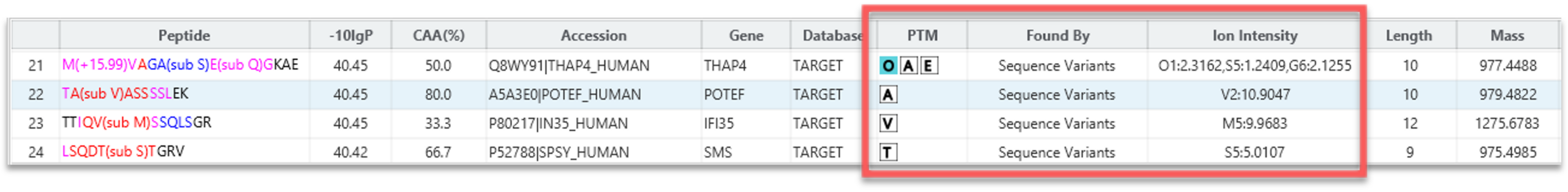
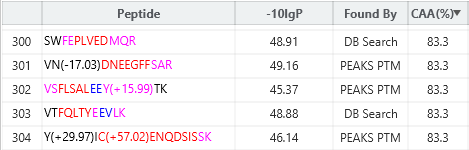
- Added Found By filter to distinguish peptide identifications.
- Introduced a versatile new tool in the Tools menu that enables easy generation of transition and mass lists from PEAKS results or in-silico digestions—supporting a wide range of targeted proteomics applications.
- Introduced a Labelled PRM workflow supporting SILAC-based labelling for enhanced accuracy in targeted quantification.
- Added support for timsTOF PRM Quantification.
- PRM Quantification calibration groups optional.
- Added PRM protein inference with protein page and visualisation.
PEAKS®: Advanced Proteomics Solution
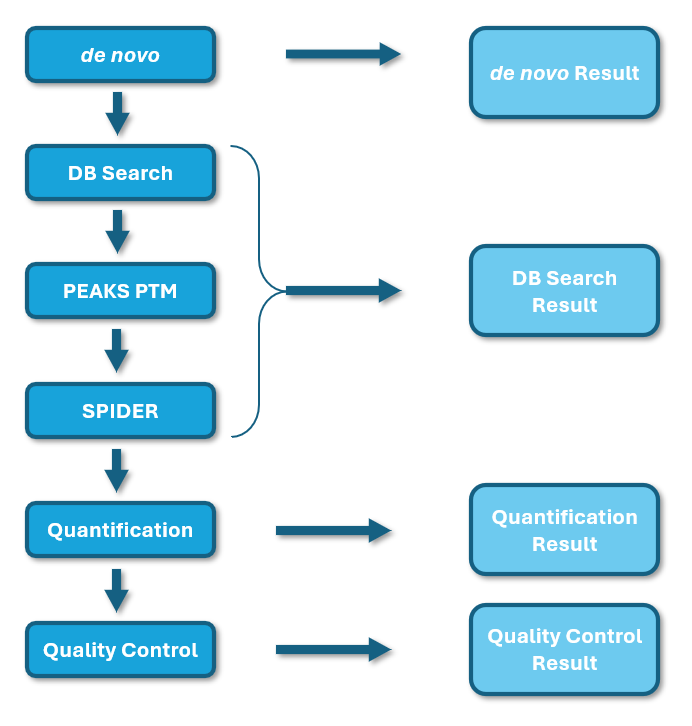
PEAKS® is a specialised tool that offers unrivalled peptide identification rates driven by deep learning technology integrated with library search, direct database search and de novo sequencing. PEAKS® 13 supports data-dependent and data-independent acquisition analyses (DDA and DIA respectively) and ion mobility mass spectrometry (IMS-MS) with the IMS add-on module. As a vendor-neutral computing platform, PEAKS® is capable of directly loading raw mass spectrometry data and standard data formats. Deploy the PEAKS® workflows to identify the presence of peptides and proteins in your project for 1) DDA data analysis including de novo sequencing, PEAKS DB (database search) identification, PEAKS PTM (post translational modification) analysis, SPIDER homology search and PEAKS DeepNovo Peptidome (specialised workflow for peptidomics data) and 2) DIA data analysis including PEAKS DIA Workflow (spectral library search, direct database searching and de novo sequencing),and DeepNovo-DIA Peptidome.
Quantification analysis by labelling and label-free quantification (LFQ) can also be performed using the PEAKS Q add-on module. Intuitive result visualisation tools are provided at every stage of analysis and results can be exported. Targeted data analysis includes PEAKS PRM Quantification and Hybrid-PRM/DIA.
Newly Redesigned and Streamlined DDA and DIA Proteome and Peptidome Workflows


Workflows with common steps are now consolidated into the same workflow. Users can choose which point to finish the analysis and can easily add additional steps later.
Deeper Discovery with Sequence Variantsuence Variants
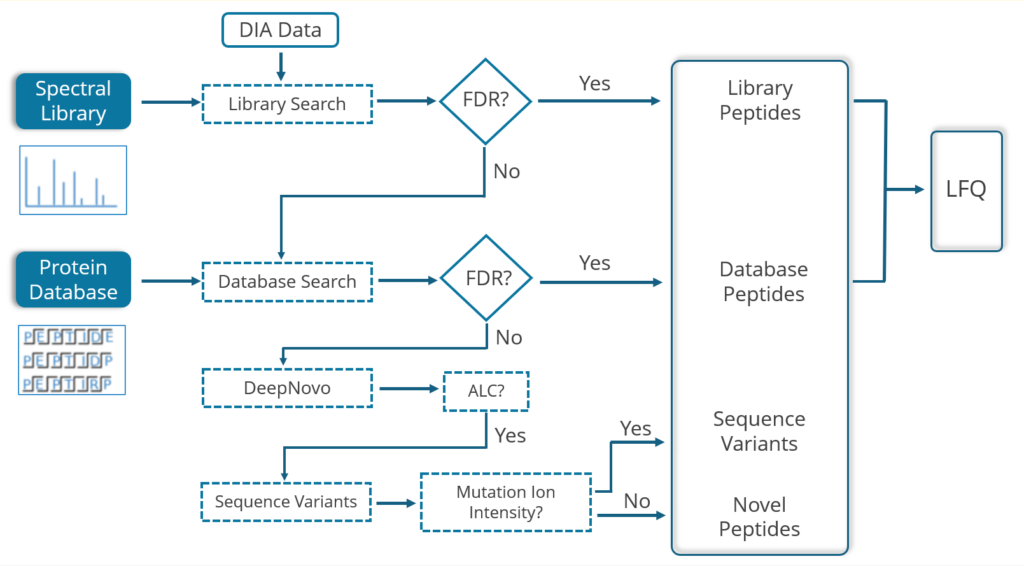
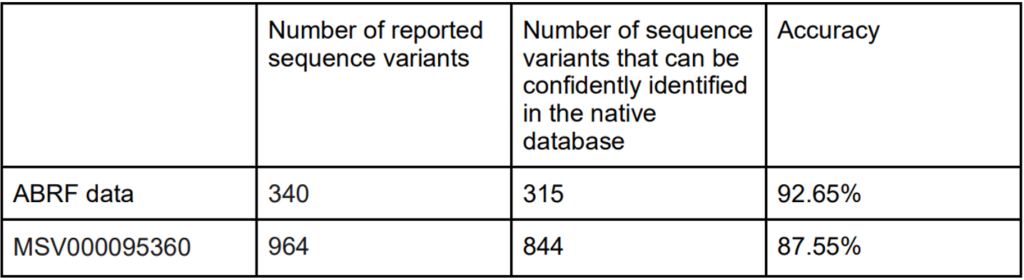
PEAKS 13 DIA can accurately identify sequence variants and novel peptides from a human dataset via a simulation test where an incorrect mouse database was used to search the human dataset. PEAKS could identify the correct human peptides through sequence variants compared to the mouse database and through the identification of novel peptides [1].
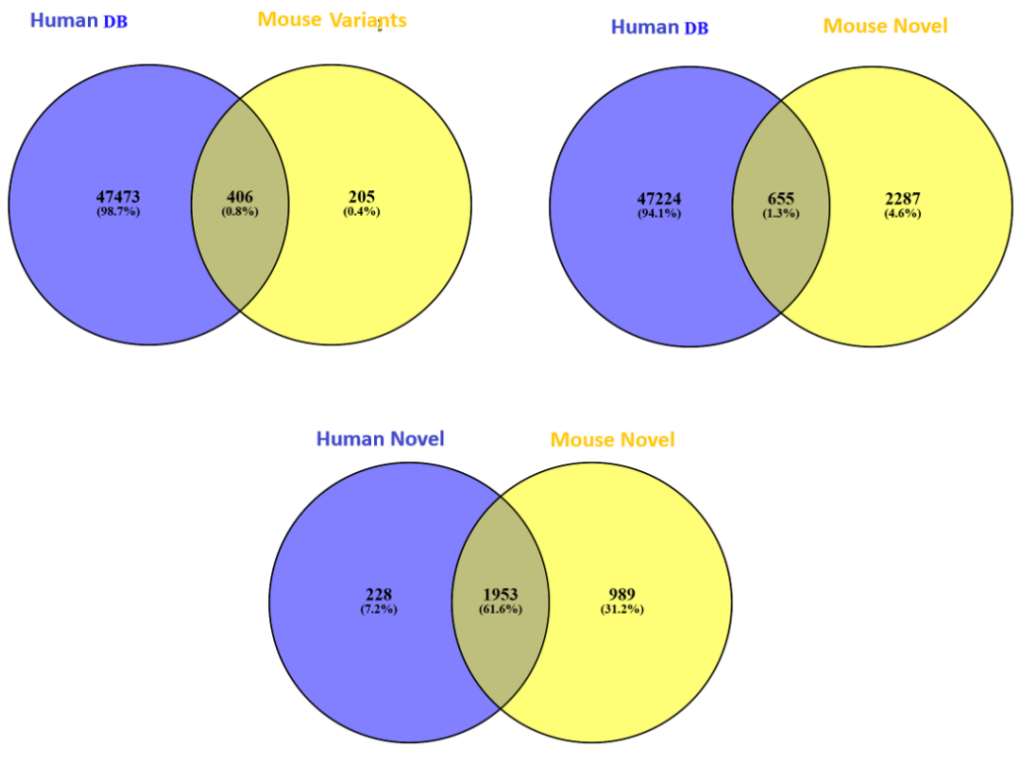
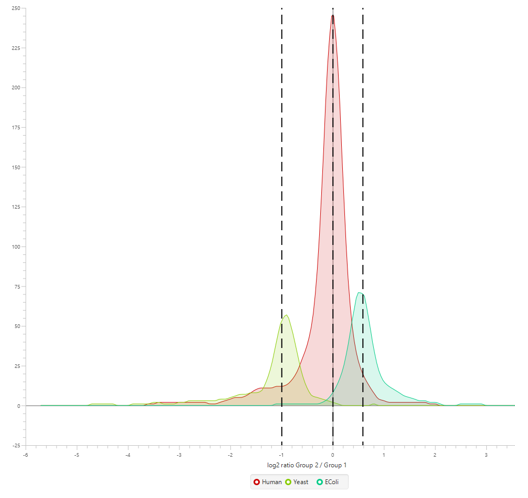
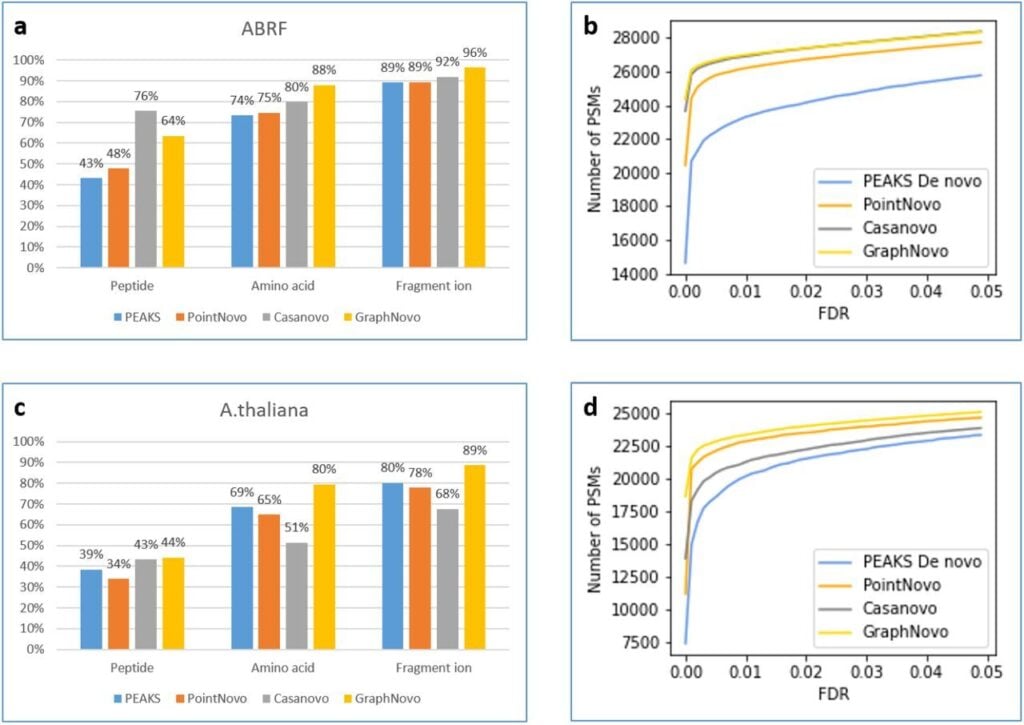
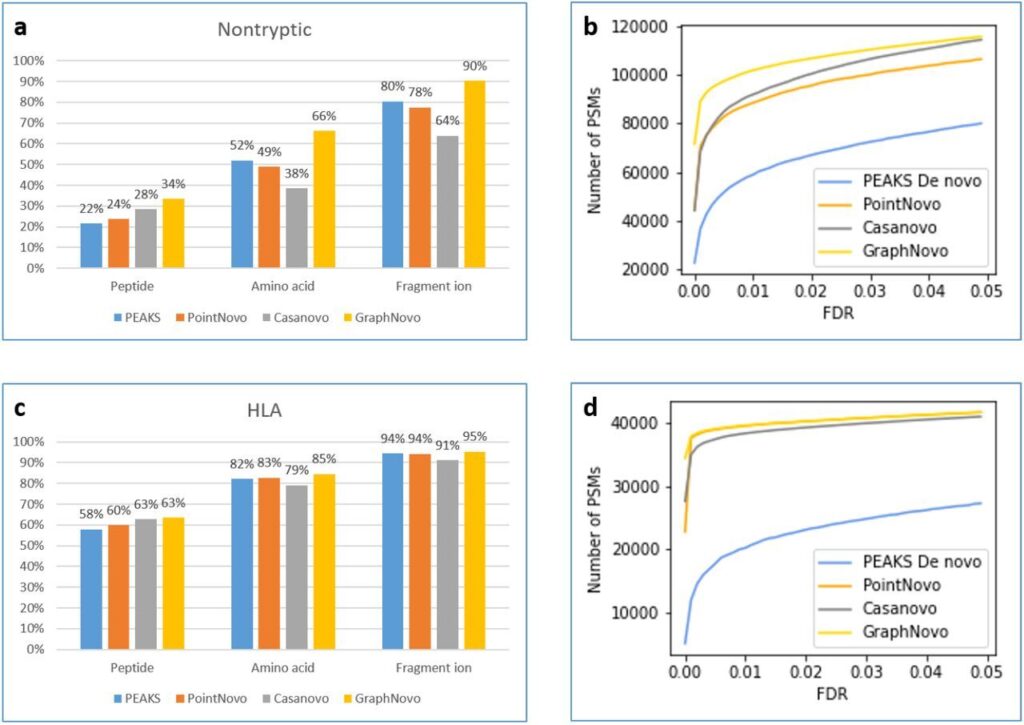
Furthermore, in our latest study [2] we demonstrate the accuracy of our new DIA solution for increased depth through cross-species validation test, addressing critical gaps in existing analytical frameworks:
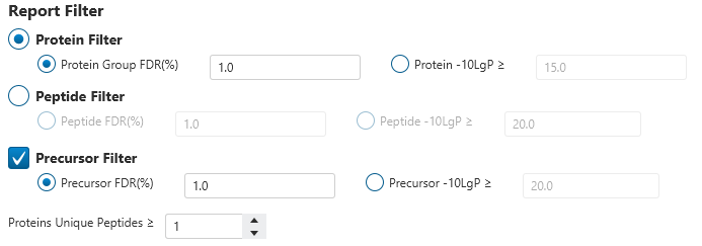
Enhanced FDR control and result filtering
Selecting report filters at both project and sample levels ensures a confident result.

Protein Quantification using Top3 or MaxLFQ
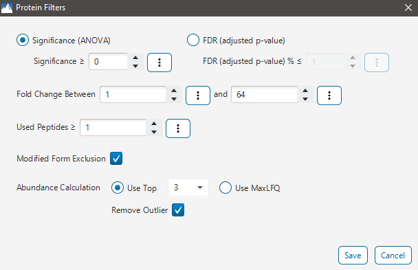
Users can now select the quantification algorithm used in PEAKS DIA LFQ based on their research needs.
PEAKS DIA Peptidome with Sequence Variants
DIA Peptidome incorporates the powerful deep-learning based GraphNovo for peptide sequencing and sequence variant analysis for increased and more confident identifications [3].
LFQ use both Database found peptides and Sequence Variants.
DDA Proteome now allows for
labelled and label free quantification for PEAKS PTM and SPIDER result. Intuitive results nodes combine the results from all steps of the search.
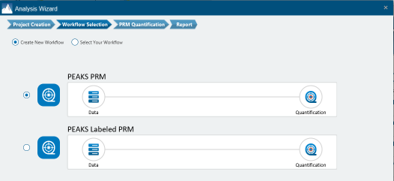
More flexible Targeted Proteomics workflow
PEAKS targeted proteomics workflow is optimised for more flexibility and ready-to-use purpose. The workflow is separated into unlabelled and labelled workflow for different application scenarios. PRM PASEF (Bruker) is now supported in PEAKS 13.
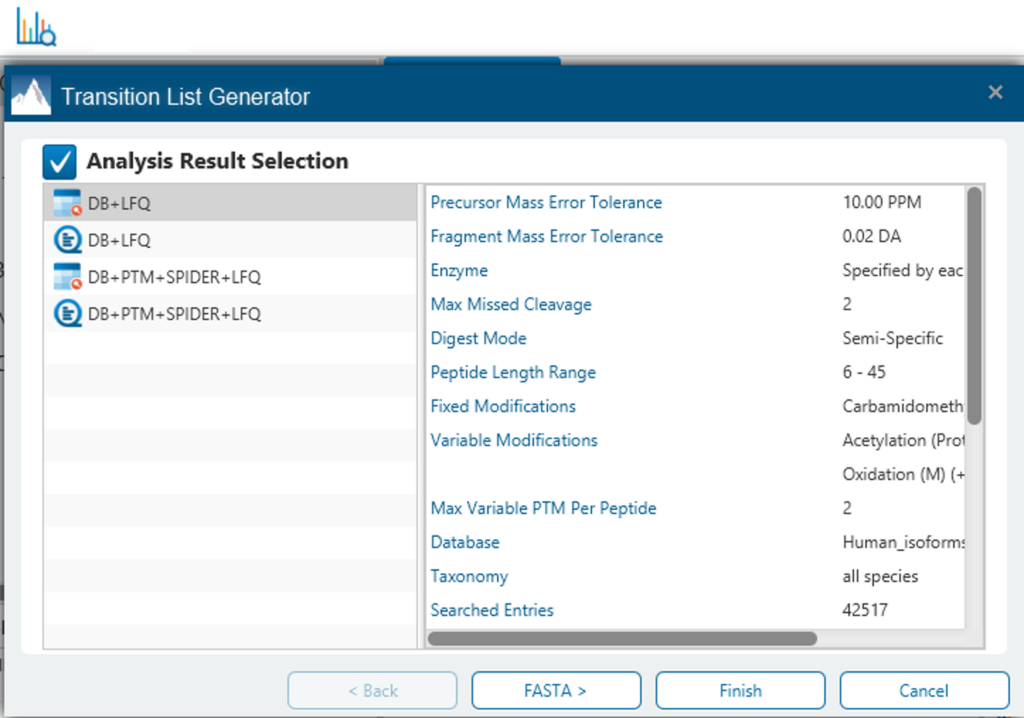
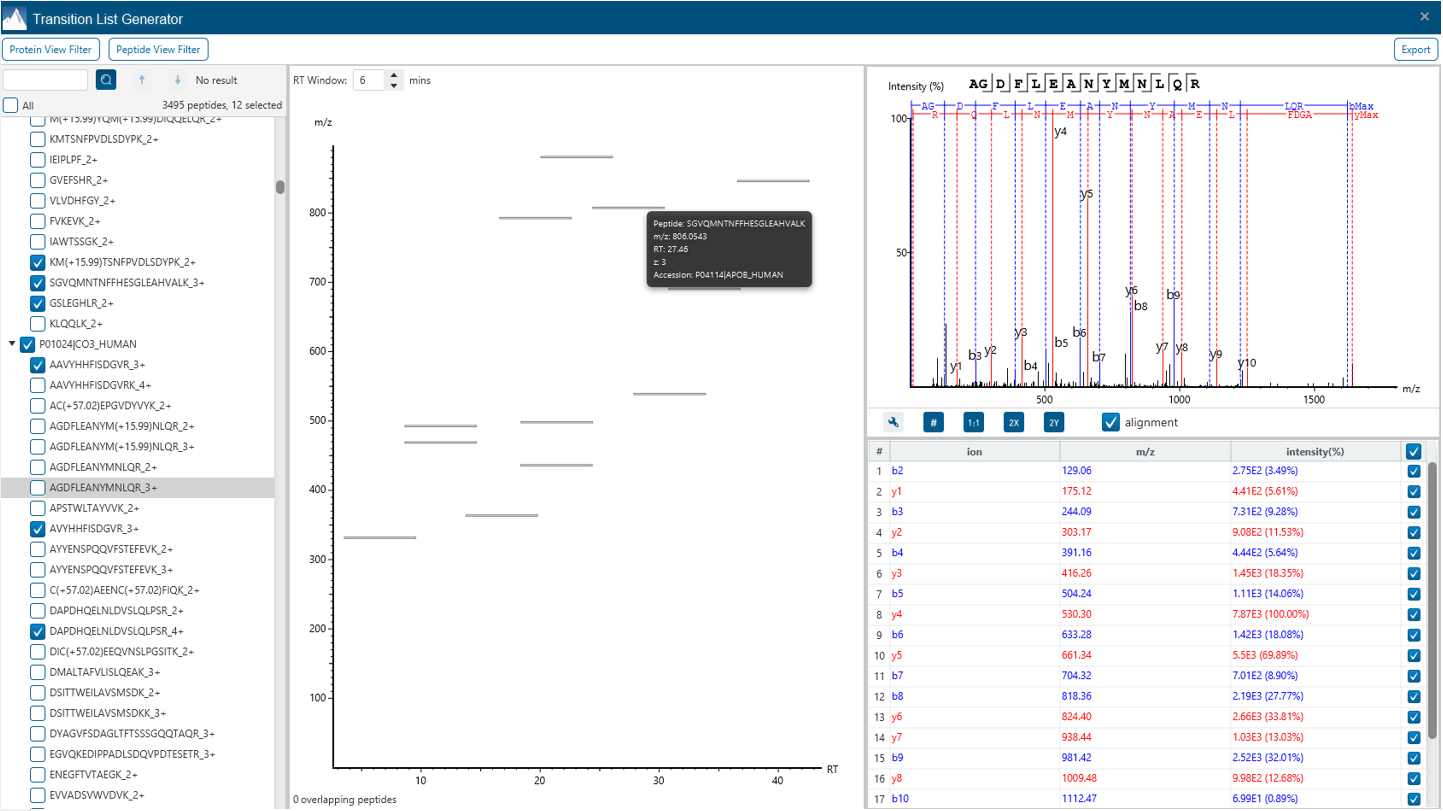
Transition List Generator for easy selection of your target peptides
The new Transition List Generator tool is implemented as a ready-to-use interference between discovery proteomics and targeted proteomics. User can select/visualise fragment ions and scheduled transitions.
References & Resources
References
- Shan, B., Korkola, N. Uncover the hidden peptidome/proteome with sequence variants and novel peptides from DIA data. Bioinformatics Solutions Inc., Waterloo, Canada [Download Link]
- Qiao, R., Li, H., Bian, H., Xin, L., Shan, B. De Novo sequencing-assisted homology search for DIA data analysis enables low abundance peptide variants discovery. bioRxiv (2025). doi:10.1101/2025.05.30.657054
- Mao, Z., Zhang, R., Xin, L. et al. Mitigating the missing-fragmentation problem in de novo peptide sequencing with a two-stage graph-based deep learning model. Nat Mach Intell 5, 1250–1260 (2023). doi:10.1038/s42256-023-00738-x