

PEAKS AB 3.0 Software New Features
PEAKS AB automates protein de novo antibody sequencing using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) datasets from orthogonal enzyme digests. From confident de novo sequence tags, the full protein sequence is assembled using a weighted de Brujin graph1.
In PEAKS AB 3.0, the software package is equipped with additional tools to enhance sequencing accuracy and result presentation, including:
- Sequence assembly of non-antibody proteins
- In-Depth Glycan Profiling
- Manual editing and finding homologous sequences
- Advanced intact Mass Deconvolution Analysis
- Support ZenoTOF EAD
In-Depth Glycan Profiling
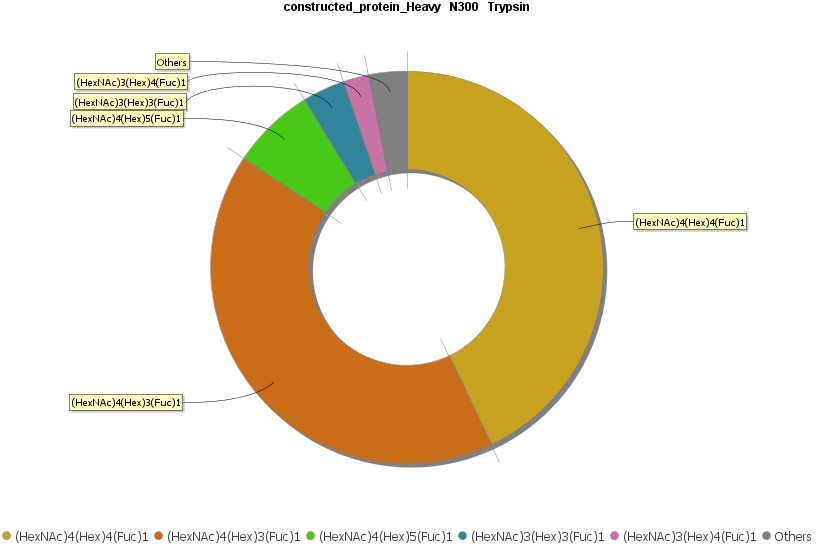
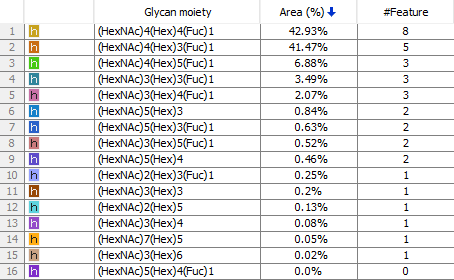
A new feature introduced in PEAKS AB 3.0 is the Glycan Profiling tool. This tool performs in-depth glycan profiling for N-linked sites identified in the heavy and/or light chains of the antibody. In addition to accurate glycopeptides mapping to the assembled antibody sequence, enzyme-based glycan profiling displays the composition and relative abundance of each glycan at a selected glycosylation site. Glycan composition and structure annotation are provided within each glycopeptide spectrum and are based on glycan fragment ions that match to an N-linked glycan database. Accurate localisation of the glycan at each N-linked site is achieved by identifying fragment ions of glycan moieties associated with the peptide backbone.
Sequence Assembly of Non-Antibody Proteins
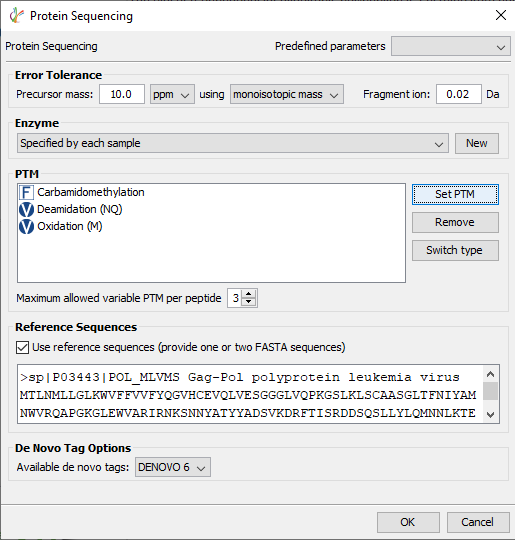
In PEAKS AB 3.0 protein samples other than antibody can be sequenced. The box next to “Use reference sequence (provide one or two FASTA sequence)” should be checked and the target sequence can be added to the white box below.
Just as in antibody sequencing, assembly of de novo tags is used to retrieve the unknown sequence, however, the reference sequence(s) provided can also be used to identify peptide spectral matches and fill sequence gaps.

Manual Editing and Finding Homologous Sequences
Sequence validation and manual editing is an essential step of any protein sequencing project. Sequencing errors can arise from background contamination of homologous proteins or from low coverage and data quality. By selecting 3 or more amino acids, the user can now select “Find homology sequences” to reveal any alternative peptide sequences that could map to that region.
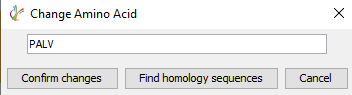
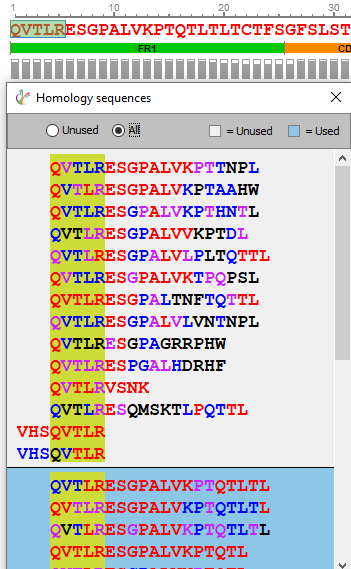
The alternative peptide sequences displayed are colour coded based on local confidence scores for each amino acid, for quick assessment of peptide sequence quality. By selecting the amino acids to be edited, then changing to the corrected sequence, clicking “Confirm changes” will edit the reference sequence. Applying these changes from the Peptide Mapping window will generate a new sequencing result node after a second round of sequence assembly.
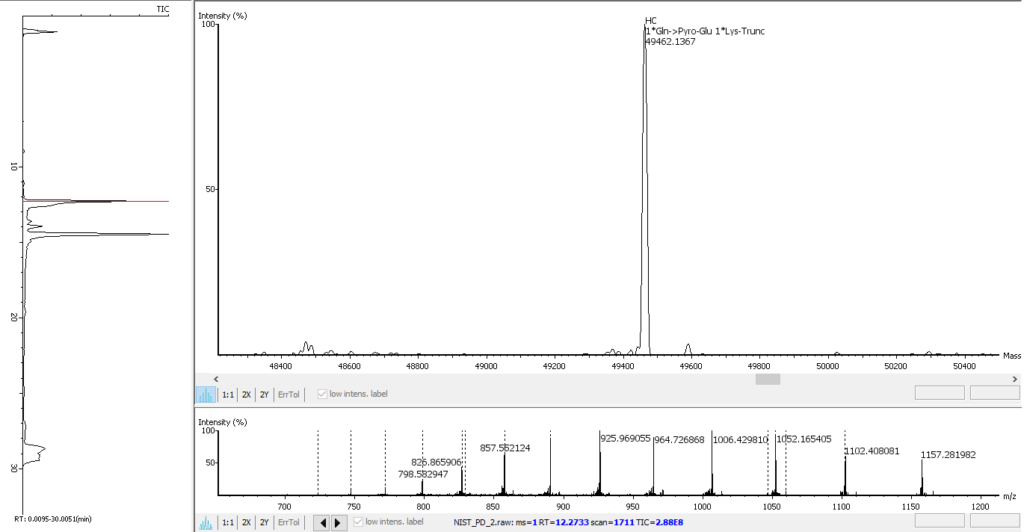
Advanced Intact Mass Deconvolution Analysis
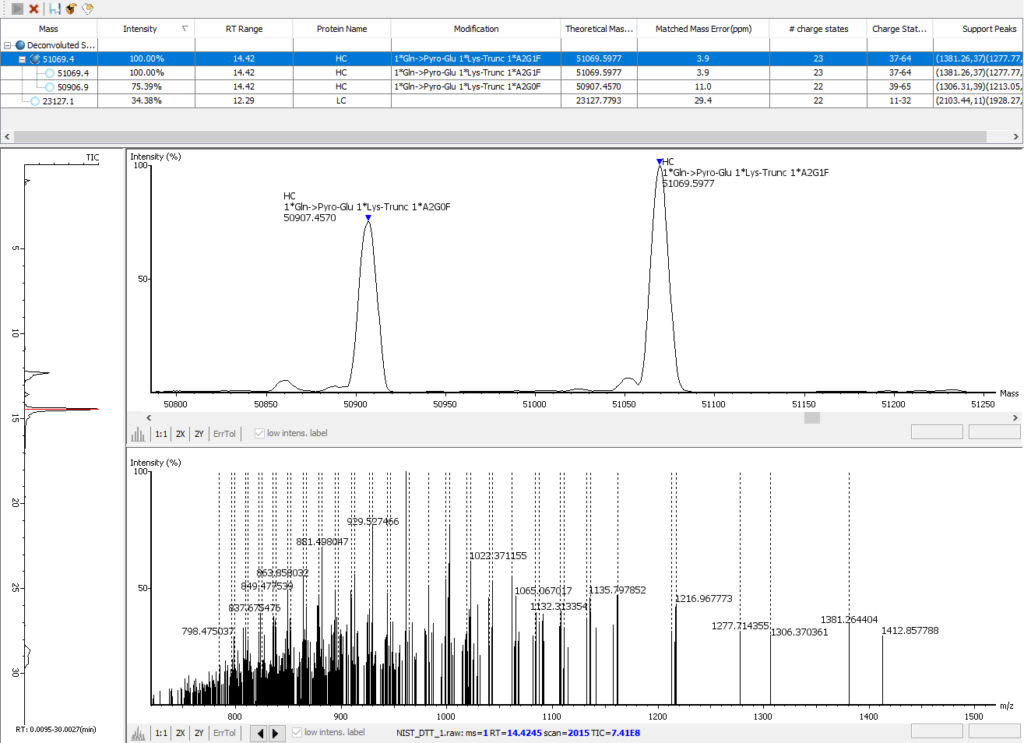
The PEAKS AB 3.0 advanced deconvolution algorithm enables automated, efficient, and accurate intact mass analysis. Intact mass measurements of proteins are performed to validate the assembled sequence and presence of any modifications such as N-terminal pyro-glutamate (Gln->Pyro-Glu), C-terminal lysine truncation (1*Lys-Trunc), and N-linked glycosylation (1*A2G0F), as shown below.
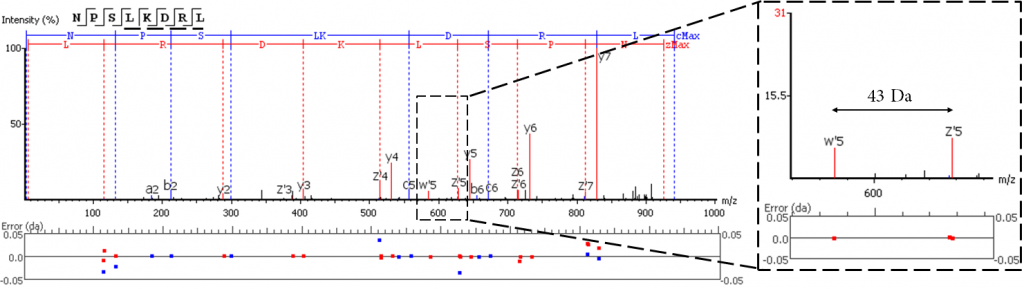
Enhanced 3-Tier Ile/Leu Differentiation
EThCD or EAD fragmentation produces signature w-ions from the characteristic loss of -C2H5 (-29 Da) from isoleucine and -C3H7 (-43 Da) from leucine. PEAKS AB 3.0 uses this information, plus enzyme digestion specificity and homolog database analysis for improved Ile/Leu discrimination.
- z ion with I on its N-terminus loses ethyl (-29)
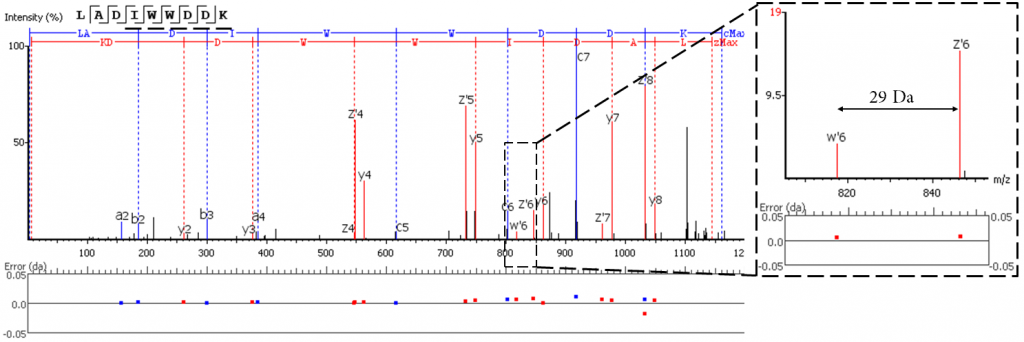
- z ion with L on its N-terminus loses isopropyl (-43)
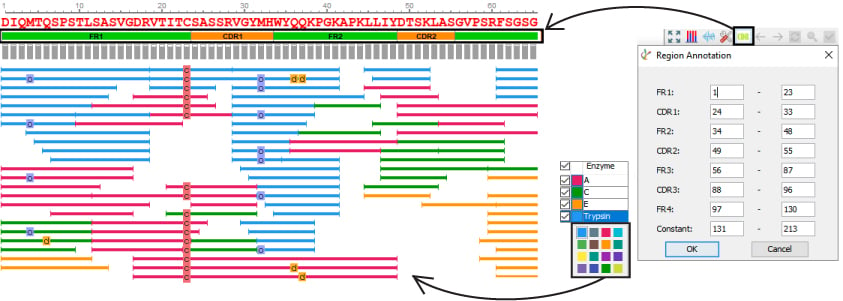
Greater Customisation Capabilities
PEAKS AB is designed to be user friendly and ultimately reduce the amount of manual work required for protein de novo sequencing. Accessing the results visually provides the user additional advantages to examine their results with precision.
- Customise enzyme colours
- Automated and customisation CDR annotation
References
- Tran NH, Rahman MZ, He L, Xin L, Shan B, Li M. Complete De Novo Assembly of Monoclonal Antibody Sequences. Sci. Rep., 6, 31730 (2016). https://doi.org/10.1038/srep31730