

Why sequence a monoclonal antibody?
Monoclonal antibodies (mAb) have a wide range of applications, from research use to the development of novel therapeutics. Isolating a mAb of interest starts by triggering its production in response to a particular antigen. Typically, an animal is exposed to the antigen then a hybridoma cell line is made by fusing the mAb producing cells with a myeloma cell line. Individual hybridoma clones are then isolated and screened for antigen specificity. This process can be both time consuming and expensive. To bypass the requirement of animals and hybridoma cell lines for mAb production, a mAb can be recombinantly expressed if the protein sequence is known. Furthermore, recombinant expression of a mAb is highly reproducible and scalable in a cost-effective manner, resulting in consistent activity from batch to batch. PEAKS AB de novo Antibody Protein Sequencing Service provides the answer to this.
Key Features:
- FAST: 1-2 week turnaround time
- Full and In-depth Sequence Coverage: Each amino acid is mapped to more than 20 unique peptides and 100% sequence coverage
- Accuracy: Every amino acid in CDRs is confidently supported by pairs of intense fragment ions in at least 10 MS2 scans
- Validation by Intact Mass: Intact mass measurements of heavy and light chains for double-confirmation of C-terminal lysine truncation and assembled de novo sequences
- 3-Tier Leu vs. Ile Differentiation: Differentiation of isoleucine and leucine using advanced EThcD MS method, enzyme digestion specificity and homology database analysis
- Interactive Viewer: Investigate details of the report directly from the PEAKS AB software GUI
Request Pricing / Schedule Consultation

$5,000/sample for New Customers
New to our sequencing services?
Sequence samples* for only $5,000 USD/sample on your first order!

Batch Sample Discounts
Have a big project?
We offer discounted pricing for multi-sample orders to ensure an affordable project.

We love your feedback!
Service customers receive a 5% discount for their next order for writing us a review, and 10% discount for every referral.
* Samples must be either a monoclonal antibody or purified, target protein for new customer pricing.
What do we provide?
Bioinformatics Solutions Inc. offers cost-effective antibody sequencing services with fast turnaround times (1-2 weeks). As a Software company and a Contract Research Organization, we can perform all sample preparation and complex mass spectrometry experiments in-house using our state-of-the-art equipment and facility. The data is then analysed using our PEAKS AB software. Manual verification and sequence analysis is included with every experiment. Additionally, our services are individually customised and tailored to your specific needs. We offer personal consultations to develop your project, and perform all experiments in a professional, accurate, and timely manner.
Why use our service?
Bioinformatics Solutions Inc. has developed the proprietary de novo antibody protein sequencing technology, based on our PEAKS AB Software, to sequence antibodies 1,2.
The Antibody Sequencing Service consists of full length heavy and light-chain antibody sequencing for all species, isotypes and allotypes.
Numerous successful cases from our antibody protein sequencing service have confirmed 100% accuracy and 100% coverage. Satisfaction is guaranteed to meet our customers’ needs.
Submission

Digestion
LC-MS/MS
Manual Validation

Detailed Reports
Your research is important to us. We want to help you get the results you’re looking for. Contact us to schedule a free consultation to discuss your project. We will help you determine the optimal experimental setup for a fast turnaround time and with the best price.

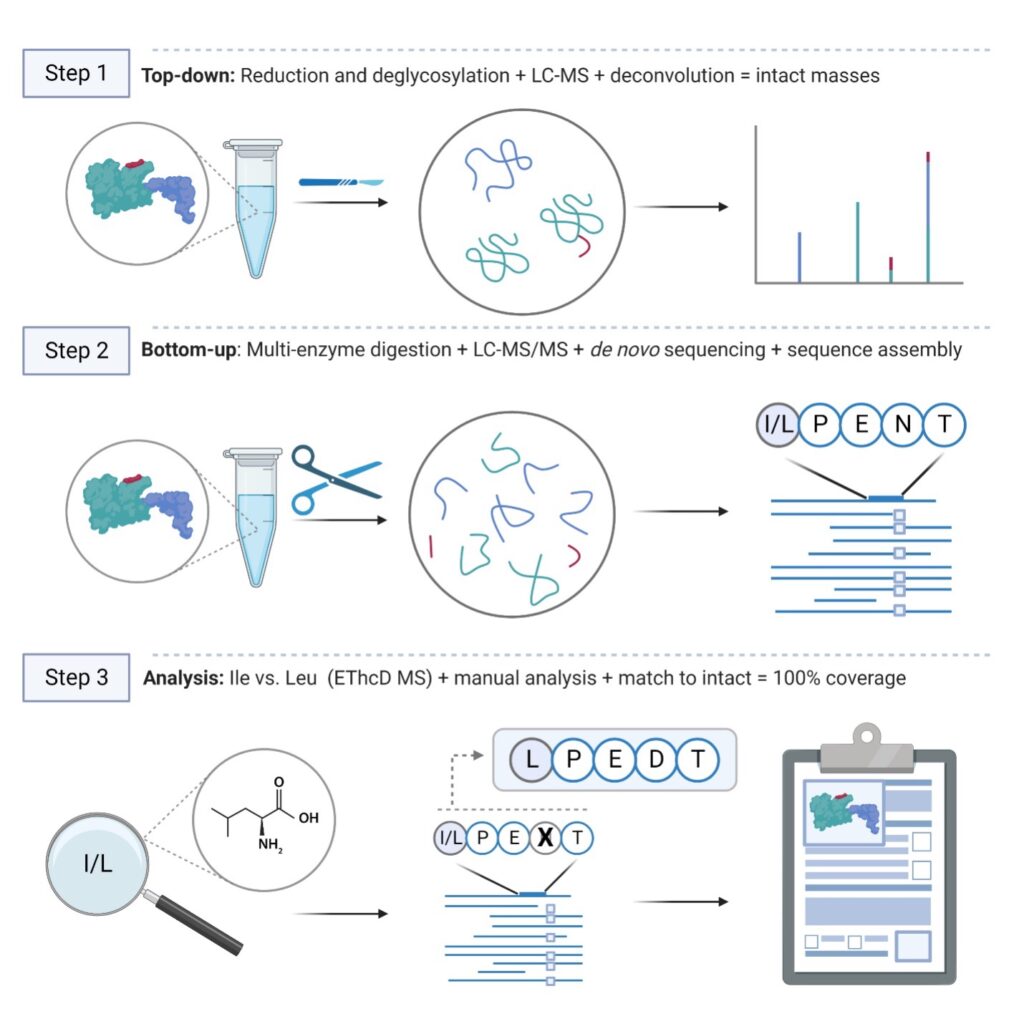
Method Overview
With over 20 years of expertise with mass spectrometry data analysis in the proteomics industry, our team keeps improving our proprietary workflow and software for de novo antibody sequencing. Our standard procedure combines top-down and bottom-up MS techniques to construct and validate the de novo protein sequence and consists of the following steps:
Service Description:
We process in-solution samples and digest samples with 5 different enzymes to obtain full protein sequence coverage. Liquid chromatography coupled to mass spectrometry (LC-MS) is used to generate raw peptide spectral data. The peptide spectral data is de novo sequenced and assembled into full protein sequences using our PEAKS AB software. Manual verification and sequence analysis are included with every project. The protein sequence is validated by matching the theoretical mass to the deconvoluted intact mass. Glycan profiling and a targeted search for post-translational modifications in PEAKS can be included as an optional add-on to this service.
Workflow:
In-solution digestion → LC-MS/MS → Protein sequencing and PTM analysis
Deglycosylation → LC-MS → Intact mass deconvolution
What we need:
- 100μg of sample provided in-solution
- Samples must be free of detergents and polymers (Triton X-100, Tween-20, PEG, etc.)
- Buffer information
Sequencing Data

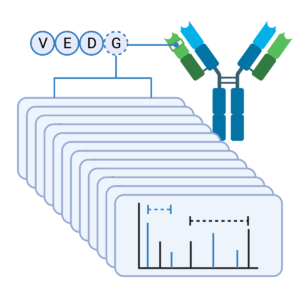
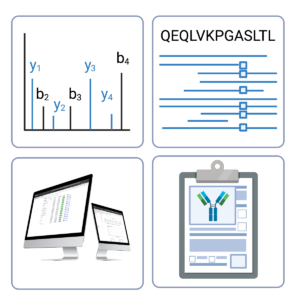
100% Sequence Coverage
Our Antibody Sequencing Service guarantees each amino acid is typically mapped with more than 20 distinct peptides and 100% sequence coverage. Data from our PEAKS AB Software will show peptides identified from the MS/MS data for each enzyme (coloured bars) or a de novo only peptide (grey). The red text of the sequence represent the confidence level for each individual amino acid (>95%) and the boxes represent identified post-translational modifications (PTM).
100% Sequence Accuracy
The Antibody Protein Sequencing Service carries out quality control from the protein to peptide and all the way down to the amino acid level based on the de novo sequencing result. Direct fragment ion evidences from MS/MS data are required for each amino acid in the assembled protein sequences. Every amino acid in CDRs is confidently supported by pairs of intense fragment ions in at least 10 MS2 scans.
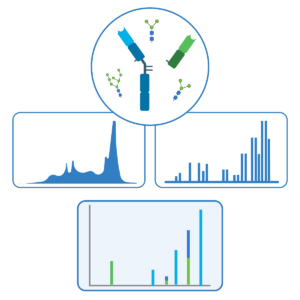
Intact Mass Validation
Intact mass measurements of heavy and light chains are performed for double-confirmation of the assembled de novo sequences, N-terminal pyro-glutamate modification, heavy chain C-terminal lysine truncation and N-linked glycans.
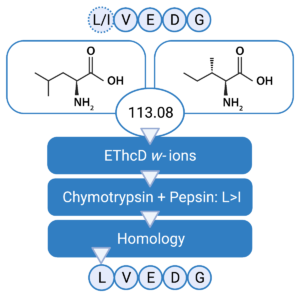
Confident Leucine & Isoleucine Differentiation
Leucine (Leu) and Isoleucine (Ile) residues are generally considered to be indistinguishable by MS. Due to this ambiguity, it is difficult to differentiate between the two residues and this can impose serious consequences on the overall performance of the antibody’s specificity and affinity. In our Antibody Sequencing Service, we use an integrated strategy that combines 1) w-ion detection in EThcD, 2) enzyme cleavage preference, and 3) homology statistics for unambiguous discrimination of Leu/Ile residues.
Custom Deliverables and Interactive PEAKS Viewer
The assembly of your results will be customised to your project, but may include:
- Antibody sample descriptions and detailed experimental procedures
- Complete sequence of your antibody
- Intact Mass with supporting MS data
- Peptide Mapping at 1% of FDR at Peptide-Spectrum Match Level with with confidence levels for each amino acid and identified post-translation modifications
- Typical peptides selected for variable regions and a list of abundant peptides
- Spectral evidence for ambiguous amino acids
- Evidence and confidence level for Leu/Ile differentiation
- Interactive PEAKS AB software viewer
- Raw LC-MS data

BSI Antibody Sequencing Service Guarantee
Provided that samples meet our minimum sample requirement (100 μg sent, 80% purity), we guarantee:
- Full de novo coverage of entire variable region with at least 98% confidence at each individual amino acid, supported by strong MS/MS intensity and peptide fragmentation on at least 5 unique peptides.
- At least 50 or more unique overlapping peptides throughout the entire protein†
Unique peptides are peptides that either have different sequences, modifications, charge states, or are generated by different proteolytic enzyme digests. In cases where minimum sample requires are not met, we will strive to meet these benchmarks.
Sequence Validation: We guarantee that the average measured intact mass of reduced light and heavy chains will match the calculated average mass of the determined sequences within a ±2 Da range.
† Excludes conserved glycosylation site where coverage may be lower but glycans will be present.

Want to know more? Request more details to learn why our methodology and experience gives you better results than our competitors. Have questions? We’ll be happy to answer any of your inquiries today.