
an all-in-one software for full proteomics LC-MS/MS Data Analysis
Highlights
- Spectral library support
- New additions to PEAKS label-free quantification (LFQ)
- New quality control (QC) function
- DeepNovo integration into PEAKS de novo sequencing
- diaPASEF support
Overview
Take PEAKS X to the next level with PEAKS X+ for a full data analysis workflow of any type of LC-MS/MS proteomic data, all in one software package. In PEAKS X, users were introduced to the support of DIA and IMS data. With PEAKS X+, users have a full suite of workflows and tools for any type of data analysis.

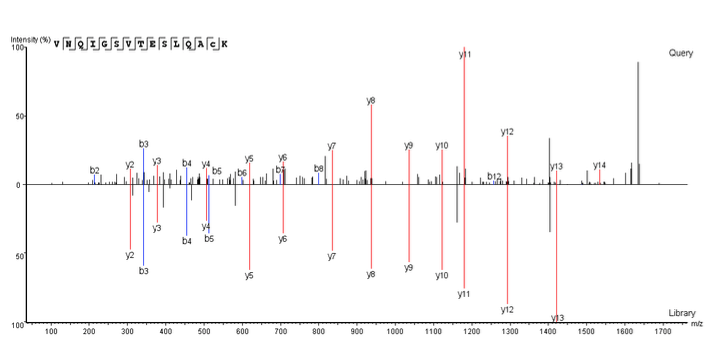
Generate reproducible, accurate identification of known peptides in DDA and DIA datasets with the spectral library searching in PEAKS X+. The new PEAKS X+ spectral library search uses m/z, intensity values, normalized retention time plus ion-mobility collisional cross section data (CCS) (with the PEAKS IMS add-on module), and is designed to work with both DDA and DIA spectral library searches. AI-based spectral prediction to improve the separation of true and false hits. With precise FDR estimation users can detect more peptides in each dataset without sacrificing accuracy for both, DDA and DIA analyses. The addition of the spectral library search to the current functions in PEAKS allows users to perform their whole pipeline in one software tool, from creating a spectral library to spectral library searching, and even de novo sequencing and database searching with the remaining scans.
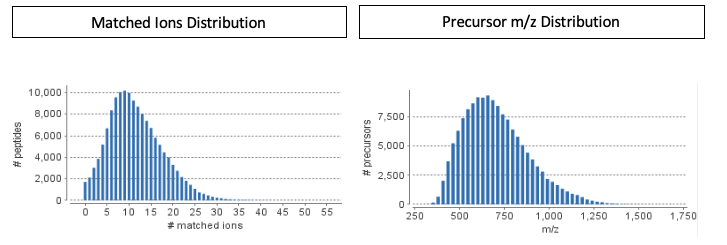
Spectral libraries generated in PEAKS are comprehensive libraries. This means that the library contains both matched and unmatched peaks, the RT information, and the associated database protein. By producing comprehensive libraries, users can easily perform further downstream analysis. Once the library is generated, configuration of the library in PEAKS Studio X+ automatically generates quality control charts for library validation
Using the spectral library, users can take advantage of the new identification workflow PEAKS X+ offers, that combines spectral library search, database search and de novo sequencing to provide an in-depth DDA and DIA analysis.
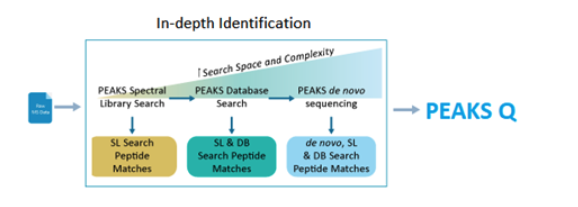
This unique pipeline provides an innovative solution to focus the analysis on peptide spectral library to gather the targeted information for the research purpose. After performing a quick screen, users can go beyond the spectral library by enabling a database search and/or PEAKS’ de novo sequencing in a single easy to use pipeline.
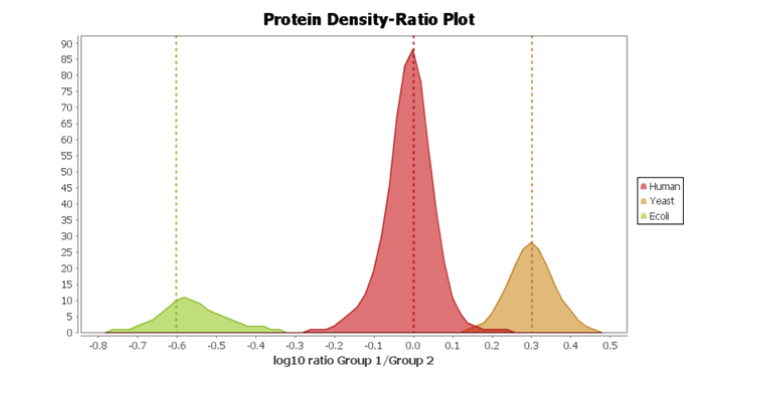
Following the new identification workflow, users can perform a quantification analysis of their data. In the example below, a SWATH dataset acquired from PXD002952 consisted of 2 samples, with 3 replicates each. The samples consisted of 3 different proteomes, which were spiked in different ratios (Human 1:1, Yeast 2:1, and E.coli 1:4). Using PEAKS Studio X+, a spectral library search and then a database search was performed, followed by a label-free quantitation assessment. Using the new Density-Ratio plot provided in the LFQ result node, users can easily visualize the actual ratio between groups where the dotted line indicates the expected ratios.
MS1 intensity-based label-free quantification (LFQ) methods are convenient, inexpensive, and accurate. PEAKS provides an unbiased LFQ method – every MS1 feature will be quantified regardless the MS2 event associated with it.
However, in some applications, the interest is only in those identified peptide features. In this case, the PEAKS Q module in PEAKS X+ provides an alternative, faster LFQ workflow – ID-directed LFQ. By directing the feature detection to concentrate on identified peptide features, PEAKS will be able to conduct accurate LFQ analysis in less time and with less memory usage.
In PEAKS X+, ID-directed LFQ is the default workflow for LFQ analyses. Alternatively, users will be given the ability to switch between the two approaches simply through the configuration setting.

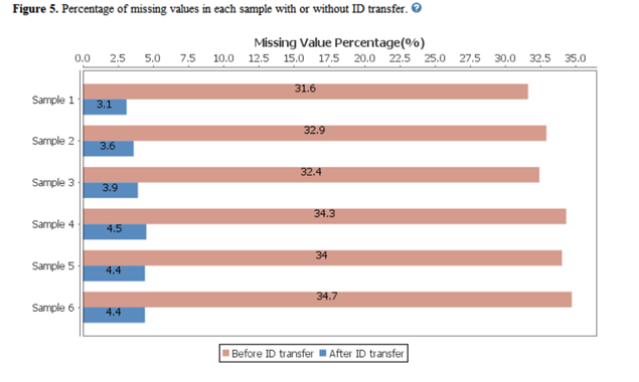
In PEAKS X+, users can now also see the efficiency of PEAKS ID-transfer between runs (also known as match between runs). ID-transfer is used to reduce the number of false missing values which are caused by the stochasticity of data-dependent acquisition (DDA) and limited quantitative depth using spectral library-based peptide identification in data-independent acquisition (DIA).
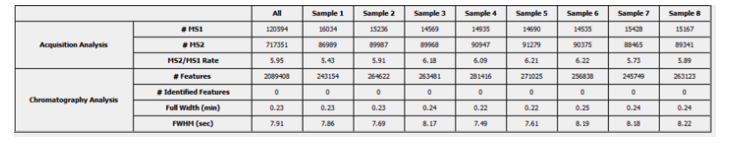
Perform a Quality Control (QC) analysis within PEAKS X+ to assess statistical information of the raw data and/or results and gain beneficial insight into the attributes of the LC-MS acquisition. This automated tool will supply the elements to determine the quality of the data and evaluate the setup of the experiment. The updated interface allows users to validate and visualize these characteristics in various tables and graphs.
- The user will be presented with a table of important statistics for both overall QC as well as individual runs.
- These sets of tables and charts will give the user an overall view of the quality of data being searched. Furthermore, it will provide a metric to compare quality of data over time to determine if there are issues with the MS or chromatography.
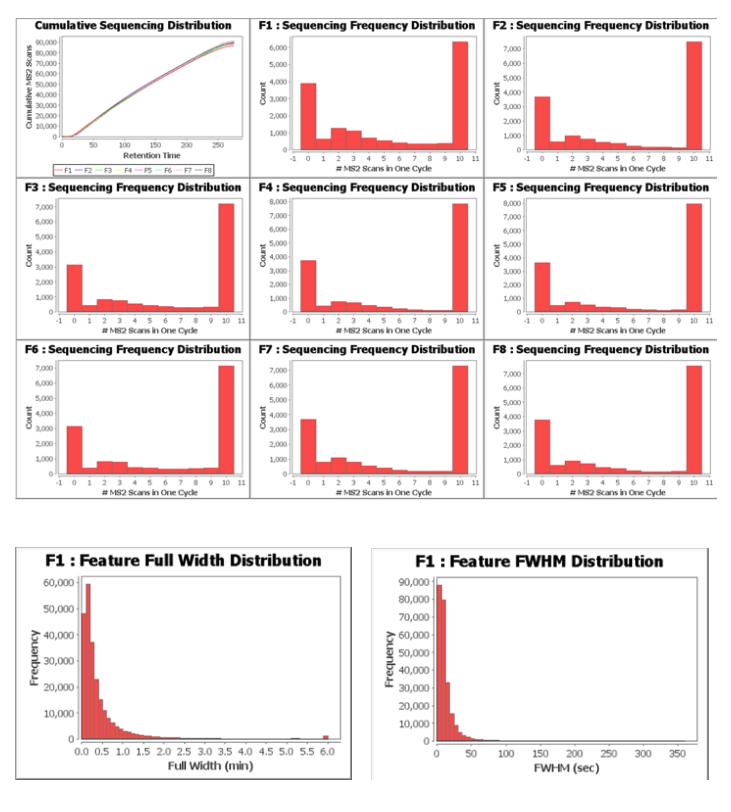
- Included in the QC metrics are graphs showing the usage of the MS in each cycle (Number of MS2/cycle – Sequencing distribution) as well as graphs showing the width of each precursor (both Full Width and Full Width Half Maximum)
PEAKS X+ integrates the proprietary deep learning-based model known as DeepNovo within its de novo sequencing algorithm. The integration of DeepNovo allows PEAKS to utilize the predicted retention time as another level of validation to resolve ambiguity between de novo peptides which have similar ion-matching scores. An example below shows how using the observed and predicted retention times improves the accuracy of de novo sequencing.
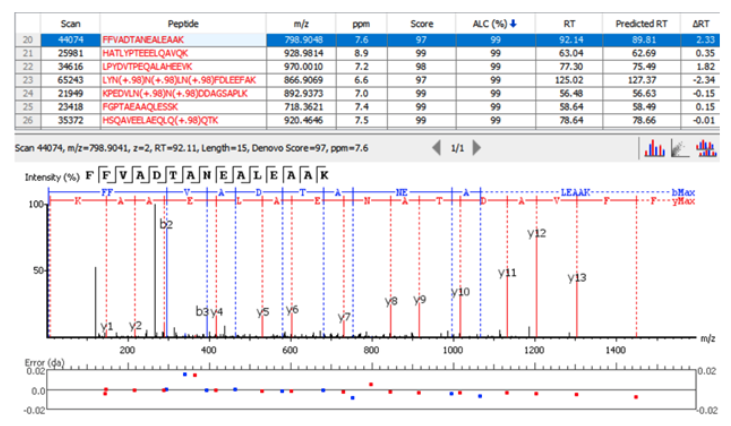
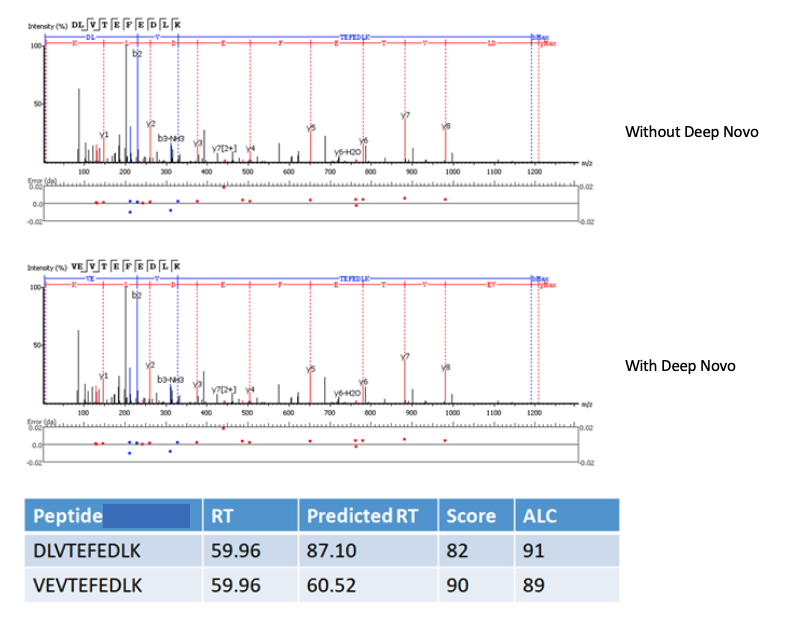
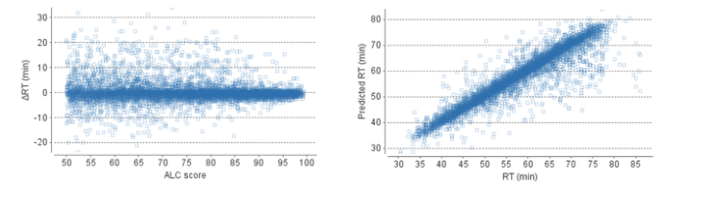
Users will find that the de novo peptide table in PEAKS X+ reports this additional information, including observed and predicted retention times, and the change in retention time (as calculated by the observed RT – predicted RT). The calculated data is also presented in scatterplots on the result summary page for users to easily assess their de novo results.
Enhance DIA experiments with the 4D capabilities of the timsTOF Pro and diaPASEF
Although parallel data acquisition leads to highly convoluted fragment spectra, the complexity can be reduced by upfront chromatography. Then by using ion-mobility as an additional separation step inside the mass spectrometer, this approach promises to reduce data complexity without a reduction in throughput. The method, called Parallel accumulation-serial fragmentation (PASEF), is available on the Bruker timsTOF Pro instrument supported by PEAKS using the PEAKS IMS add-on module.