PEAKS AB 2.0 Software New Features
PEAKS AB automates de novo antibody sequencing using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) datasets from orthogonal enzyme digests. From confident de novo sequence tags, the full protein sequence is assembled using a weighted de Brujin graph1.
In PEAKS AB 2.0, the software package is equipped with additional tools to enhance sequencing accuracy and result presentation, including:
- Upgraded de novo sequencing and assembly algorithm
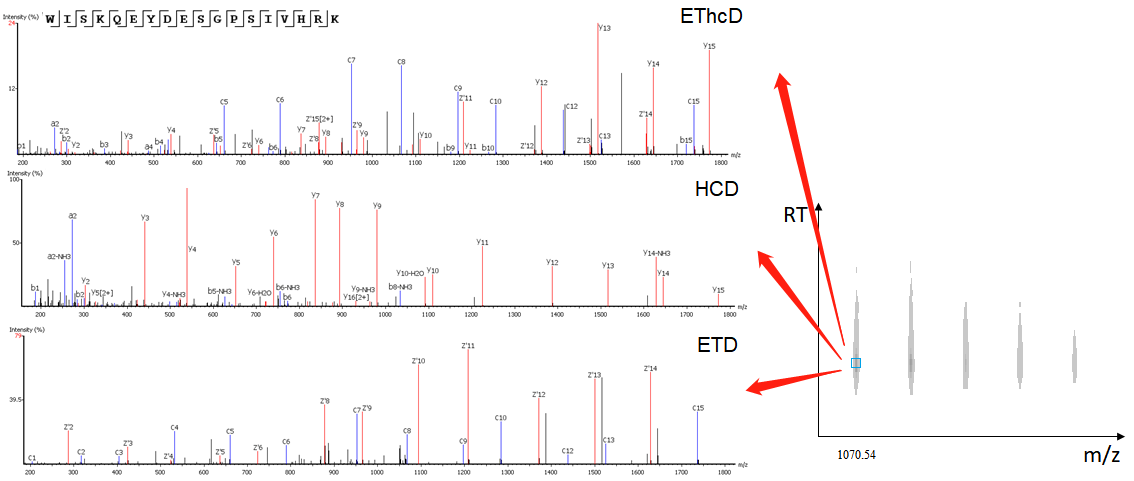
- Capability of analyzing EThcD data
- Enhanced 3-tier Ile/Leu discrimination
- Greater customization capabilities
- Intact mass deconvolution analysis
![]()
![]()
Enhanced 3-Tier Ile/Leu Differentiation
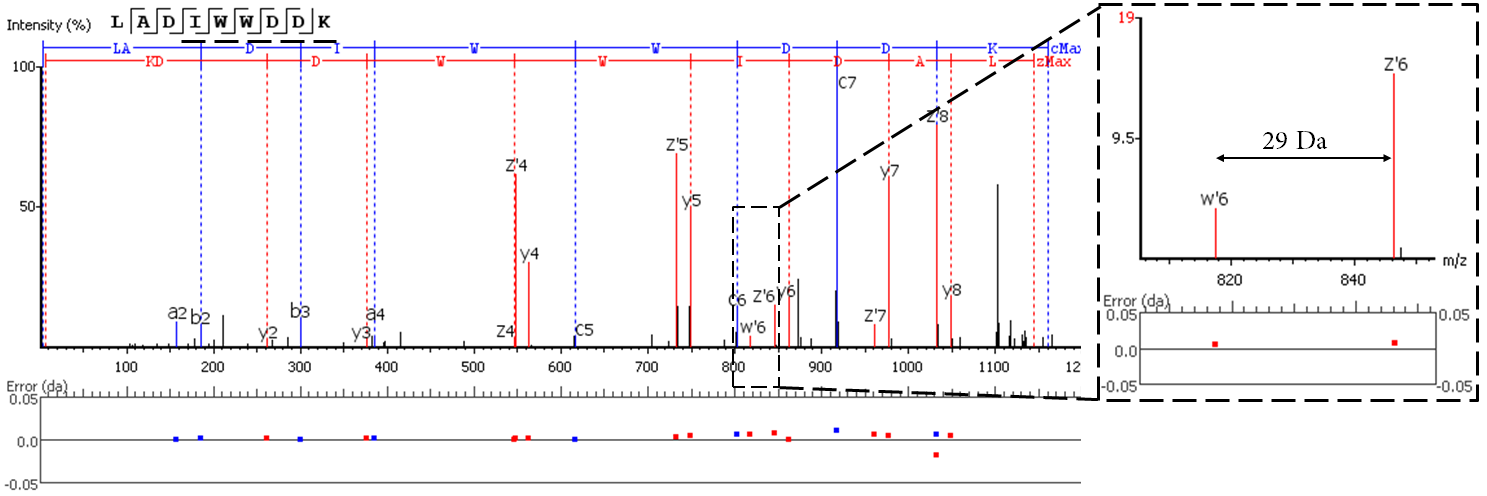
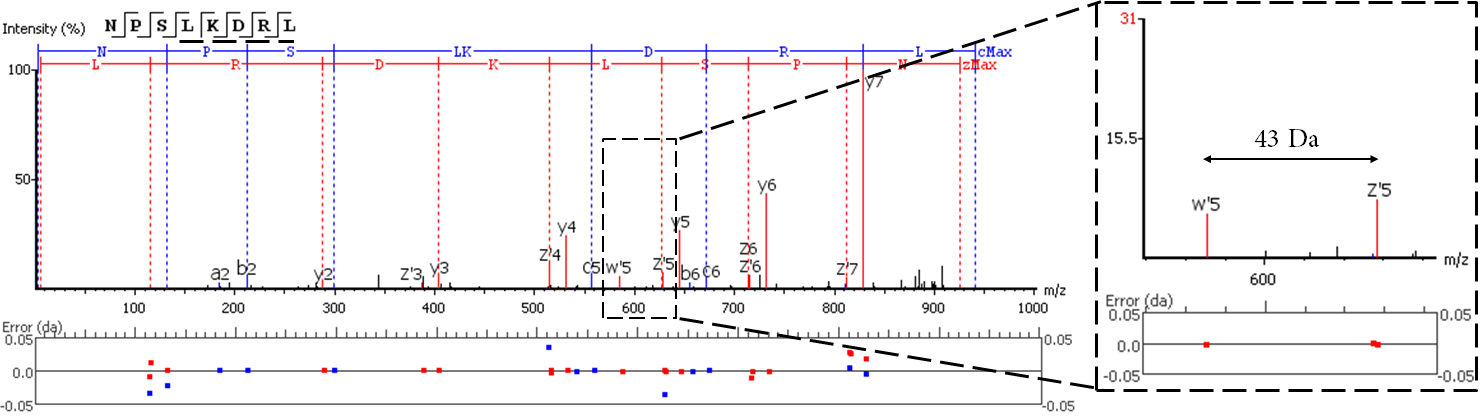
EThcD fragmentation produces signature w-ions from the characteristic loss of -C2H5 (-29 Da) from isoleucine and -C3H7 (-43 Da) from leucine. PEAKS AB 2.0 uses this information, plus enzyme digestion specificity and homolog database analysis for improved Ile/Leu discrimination.
- z ion with I on its N-terminus loses ethyl (-29)
- z ion with L on its N-terminus loses isopropyl (-43)
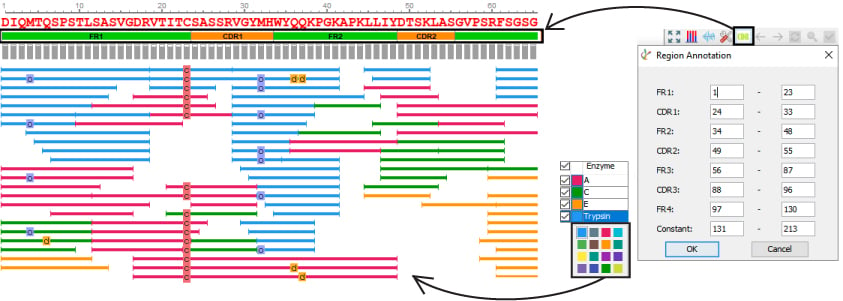
Greater Customization Capabilities
PEAKS AB is designed to be user friendly and ultimately reduce the amount of manual work required for protein de novo sequencing. Accessing the results visually provides the user additional advantages to examine their results with precision.
- Customize enzyme colours
- Automated and customizable CDR annotation
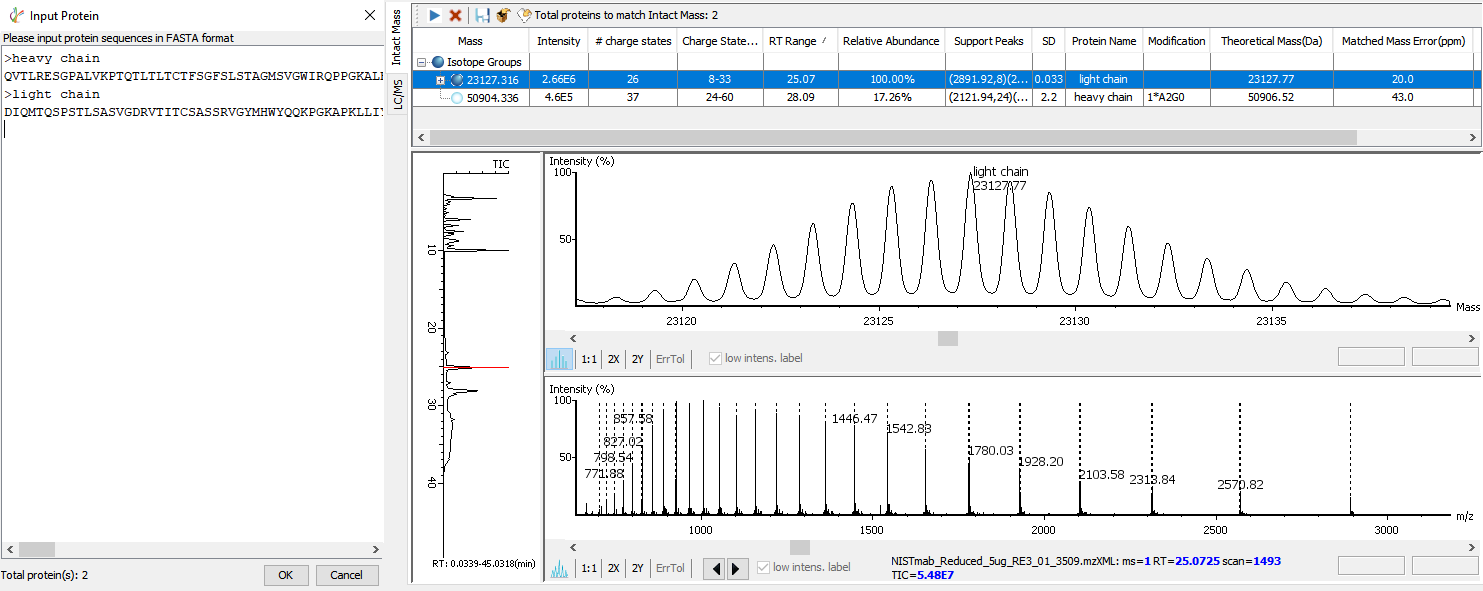
Intact Mass Deconvolution Analysis
The advanced deconvolution algorithm enables an automated and accurate intact mass spectral data analysis. Intact mass measurements of heavy and light chains can be performed for additional confirmation of the assembled de novo sequences. Some common modifications are automatically considered when matching intact mass result with the input antibody protein sequences.
Reference
- Tran, N.H. et al. Complete De Novo Assembly of Monoclonal Antibody Sequences. Scientific Reports. 6:31730. 26/08/2016.
Click here to visit the PEAKS AB Software page detailing an overview of the tool.