
 SPIDER
SPIDER
Peptide Mutations & Homology Searching
Biological samples commonly contain proteins with slightly different sequences to those protein databases. This is frequently caused by polymorphism, antibody diversity, database errors, and cross-species database searching. Ignoring those mutated peptides can potentially lead to an oversight of a potential biomarker, an error in antibody confirmation, or simply low coverage of proteins. To handle mutations, PEAKS software includes the SPIDER algorithm that is specially designed to detect peptide mutations and perform cross-species homology search.
Overview
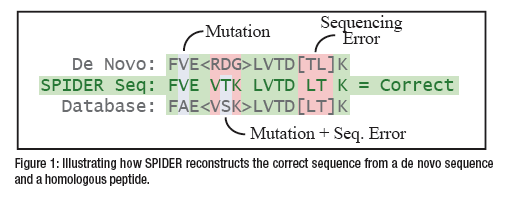
The SPIDER algorithm tries to match the de novo sequence tags with the database proteins. When a significant similarity is found, the algorithm tries to use both de novo sequencing errors and homology peptide mutations to explain the differences (Figure 1). More specifically, it reconstructs a “real” sequence to minimize the sum of de novo errors between the real sequence and the de novo sequence, and homology peptide mutations between the real sequence and the database sequence.
Why Not BLAST?
It should be pointed out that the general homology tool such as BLAST is not the best option for searching with de novo sequence tags. It is very common that some fragment ions are missing from a peptide’s MS/MS spectrum, leading to possible de novo sequencing errors. Thus, an appropriate de novo tag homology search should tolerate common de novo sequencing errors such as (AT/TA) and (N/GG). However, being designed for a different purpose, BLAST penalizes those errors too much and may significantly reduce the search sensitivity [2]. Moreover, BLAST will not attempt to reconstruct the real peptide sequence.
Besides the apparent mutation detection and cross-species search function, a very useful application of SPIDER is to use it iteratively to sequence a complete protein (e.g. antibody sequencing). This is achieved by:
- Using PEAKS’ standard workflow (de novo + PEAKS DB + PEAKS PTM + SPIDER) to search in a homologous database. This will identify a homologous protein.
- Then in the coverage pane, select tools “copy mutated protein sequence”. This will copy the mutated protein sequence (after applying the confident mutations) to Windows’ clipboard.
- Invoke another standard search by pasting the copied sequence as the protein database.
- Repeat the above procedure multiple times to gradually improve the sequence quality.
References
- Han, Y., Ma, B., Zhang, K. SPIDER: Software for Protein Identification from Sequence Tags Containing De Novo Sequencing Error. J. Bioinform. Comput. Biol., 3, 697-716 (2005). https://doi.org/10.1142/S0219720005001247
- Ma, B. & Johnson, R. De novo Sequencing and Homology Searching. Mol. Cell. Proteomics. 11 O111.01490 (2012). https://doi.org/10.1074/mcp.O111.014902