

High Throughput, Multi-User,
Proteomics LC-MS/MS Analysis
Software
PEAKS Online 11
High-Throughput, All-in-One Software for Proteomics LC-MS/MS Analysis
Use PEAKS® Online 11 to take advantage of powerful and shared computing resources to perform LC-MS/MS protein & peptide identification and quantification analyses. The restructured platform allows large datasets to be processed efficiently by multiple users at the same time; with the ability to run on any cluster, multi-CPU machine, or cloud server.
Contact us to add PEAKS Online 11 to your lab!
Highlights
- de novo sequencing.
- Database search with NEW deep learning-boost to maximise peptide ID efficiency.
- DIA workflow (Spectral Library Search + directDB + de novo).
- NEW: Library generation with de novo peptides for DIA
- NEW: Direct database search using DIA data with an improved sensitivity and accuracy including DIA with short gradient.
- Enhanced Deep learning-based technology for predicting spectra, retention time, and collision cross-section values.
- Post-translational modification (PTM) search with 500+ modification.
- Sequence variant and mutation search.
- NEW: DeepNovo-based peptidome workflow for immunopeptidomics: integrated homology search with de novo for improved HLA peptidomics with FDR control
- Label-free and Label-based: TMT (MS2, MS3) / iTRAQ, SILAC, 18D labelling, ICAT, User-Defined.
- Quantification results can be visualised using heat maps, correlation profiles, and extracted ion chromatograms (XICs).
- NEW: QC (Quality Control) – enhanced large cohort study (DDA and DIA): identification-related and quantification-related quality control measures to ensure experimental consistency, data quality and identification and quantification consistency
- Detailed and easy-to-use graphical user interface (GUI) to view, filter and validate results.
- Statistical calculations presented visually to assess quality of raw data and/or results.
- Raw data visualisation to assess total ion chromatogram (TIC), MS Scans, and MS/MS scans for each data file.
- User friendly interface to create and save workflows, search parameters, and databases for routine analyses
- True automation with PEAKS daemon: Automated Instrument Link and seamlessly connects your acquisition to your analysis in one easy to use workflow
- Align your team’s efforts with administrative controls to standardise workflows, databases, PTMs, quantification methods, and project sharing
- GPU and clustered based solution that accelerates data analysis
- NEW: PEAKS 11 now support ZenoTOF.
- Support ion mobility spectrometry proteomics (timsTOF Pro, FAIMS).
- Algorithms fine tuned for each instrument and fragmentation type to ensure optimal accuracy and sensitivity.
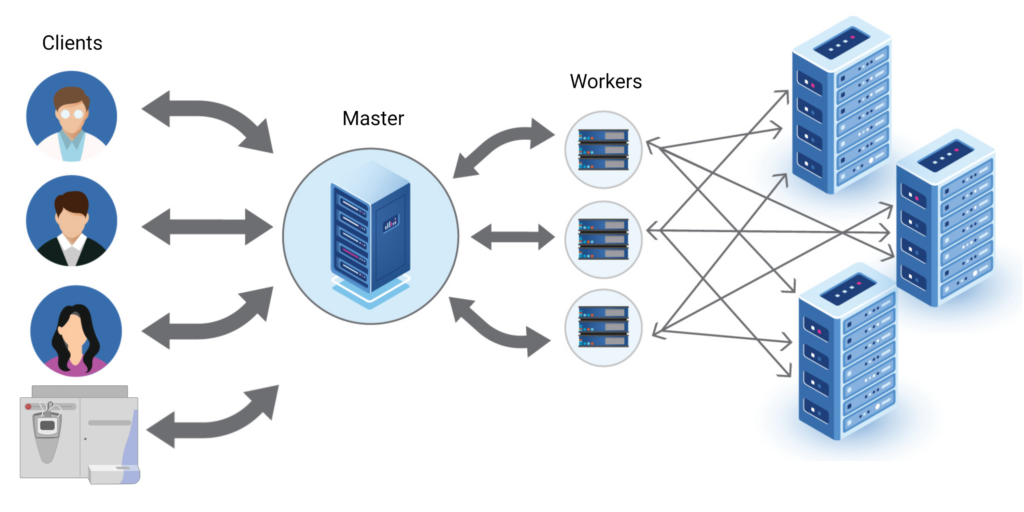
Distributed System for High performance Computing
PEAKS Online 11 uses the latest distributed computing technology to achieve high-throughput performance with established PEAKS workflows for multiple users on a network. The Master is at the centre of the system, it accepts computing request from a client or directly from the MS instrument using an automated system; PEAKS daemon: Automated Instrument Link. The Master will distribute all the computing workload to different workers, where the computation is performed. All the data which is necessary for computation will be stored in the distributed database system. Since both computation and data storage are distributed to many different nodes, single node failure will not block the whole system. Therefore, the system is always accessible to the users. Additionally, because of the distribution, the system is also highly scalable, with the ability to add additional nodes at anytime to achieve maximum performance.
Enhanced DIA Workflow: Library Search, Direct Database Search, de novo sequencing
DIA analysis is an appealing alternative to DDA approach for an increased proteins coverage. In the past, DIA methods have relied on generating spectral libraries from DDA to identify and quantify peptides.
PEAKS Online 11 offers a streamlined DIA workflow to maximise identification of peptides by integrating spectral library search and direct database search, with optional de novo sequencing.
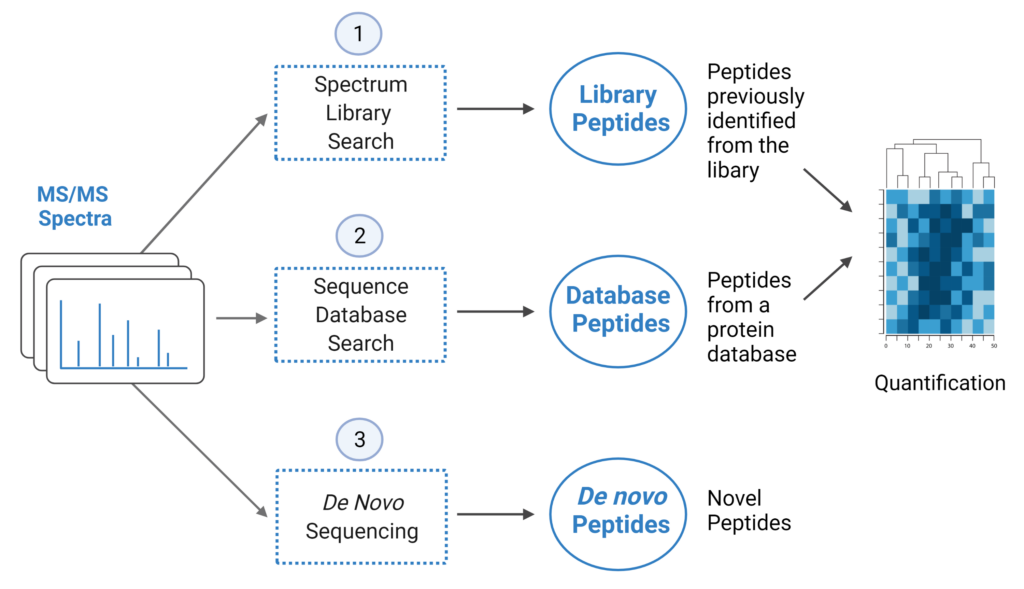
- A library search is performed against a pre-defined spectral library. Peptide spectra without a library match can be directly searched against a protein database.
- A protein sequence database is directly searched with DIA data. Advanced machine learning algorithms allow improved accuracy and sensitivity of peptide identification.
- Unmatched spectra from the database search are de novo sequenced.
- Identified peptides from both the spectrum library search and protein sequence database search can be used in a quantification analysis.
Note: All search methods, PEAKS Spectral Library Search, Direct Database Search, de novo sequencing, Quantification, and QC are optional, and the steps of the workflow can be conducted independently or in sequence.
Selecting library search followed by the direct database search results in improved protein identification rates while the completeness of proteome is further enhanced by de novo sequencing. Beside in-depth identification analysis, PEAKS Online 11 allows for quantification of DIA data by LFQ. Finally, an optional QC (Quality Control) will provide identification-related and quantification-related quality control measures to ensure experimental consistency, data quality and identification and quantification consistency.

Quality Control (QC) Function for in-depth analysis from raw data to results
With the new Quality Control (QC) analysis in PEAKS® Online 11 users can assess statistical information of the raw data and/or results and gain beneficial insight into the attributes of the LC-MS acquisition. This automated tool is designed for both, DDA and DIA data and will supply the elements to determine the quality of the data and evaluates the setup of the experiment.
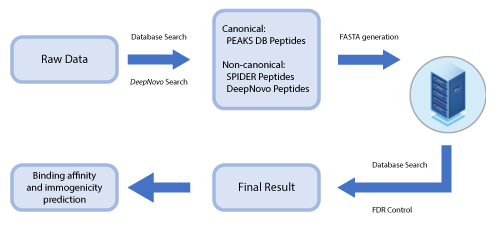
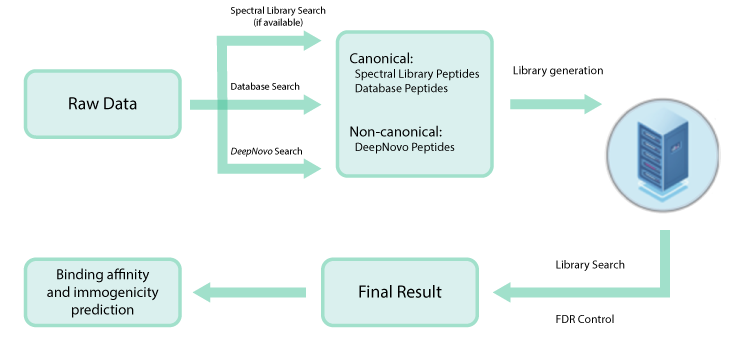
PEAKS DeepNovo Peptidome for Immunopeptidomics
This newly developed solution is a specialised workflow for peptidomics data that combines database searching, de novo sequencing, and identification of mutated peptides.
Data-independent acquisition (DIA) has emerged as a powerful strategy for deep proteome-wide identification, and PEAKS Online now enables DIA application for immunopeptidomics analyses. The new DeepNovo Peptidome in PEAKS Online is a specialised DIA workflow for immunopeptidomics and integrates spectral library, database search, and de novo sequencing. Learn more
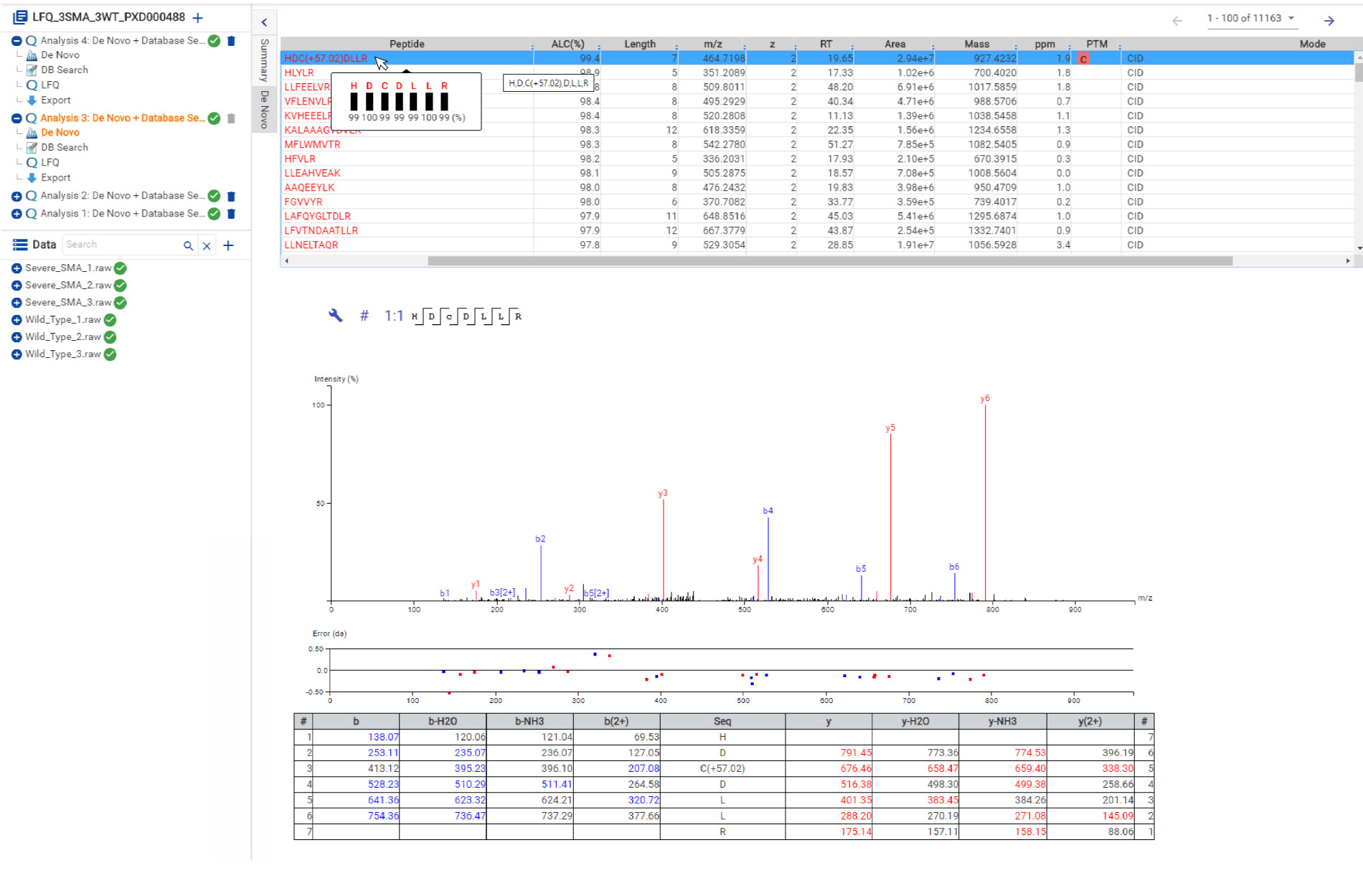
Similar to PEAKS Studio, hover over the peptide sequence to see the local confidence at the amino acid level. Each residue is also colour-coded to easily identify confident sequence tags. The local confidence score in PEAKS is the likelihood that the amino acid assignment in a peptide truly exists. The interactive annotated spectrum view, error distribution and ion match table appear below for further assessment.
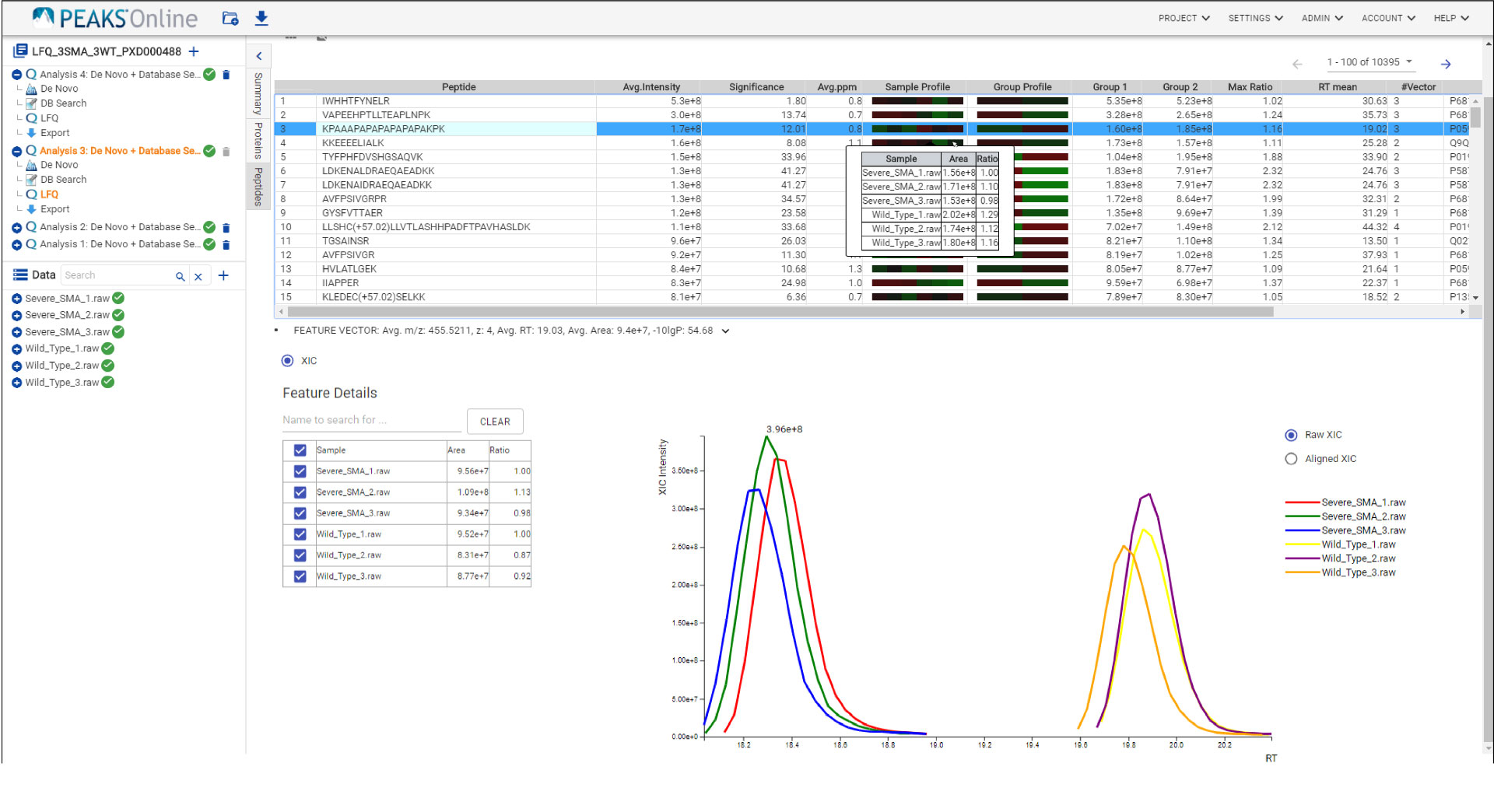
The PEAKS Online 11 interface allows projects to be analysed on all levels, from the raw data to spectrum annotation and graphical visualisation of the identification and quantification results. Result filters can be easily set and statistical tools are available every stage of analysis. With the PEAKS Q module add-on, results can be visualised using heat maps, volcano plots, and extracted ion chromatograms (XICs).
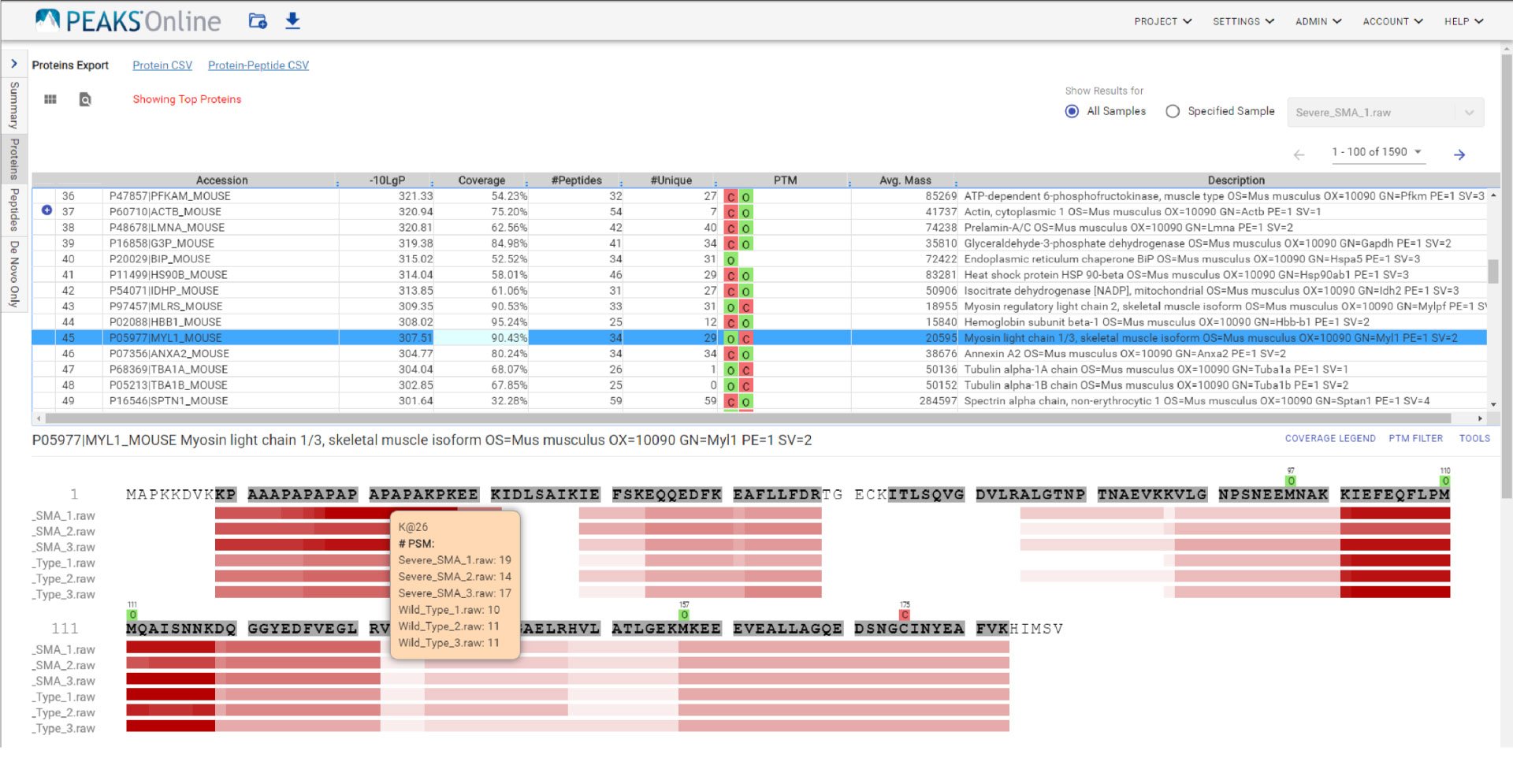
Like PEAKS Studio, the “Protein Coverage” view visually maps the supporting peptides and de novo tags to the protein selected in the Protein table. Click on a peptide of interest from the Protein Coverage view and the annotated spectrum for the corresponding peptide spectrum match (PSM) will appear. In PEAKS Online 11, the Protein Coverage view also maps the peptides on a sample basis for projects with multiple samples. This multi-sample protein coverage view allows users to quickly estimate the peptide abundance across all samples in the identification search using a heatmap.
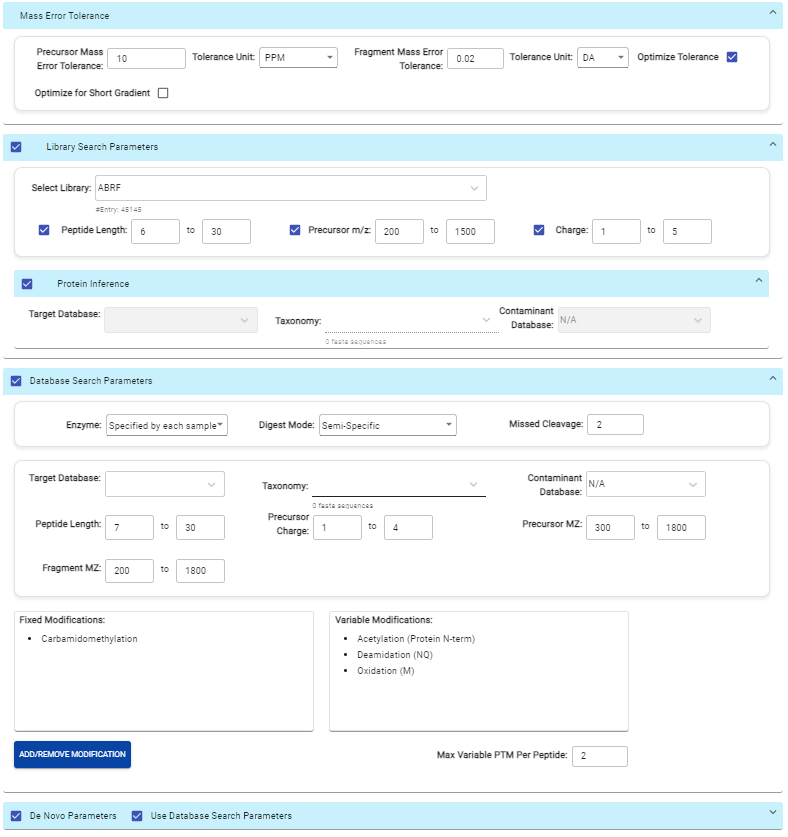
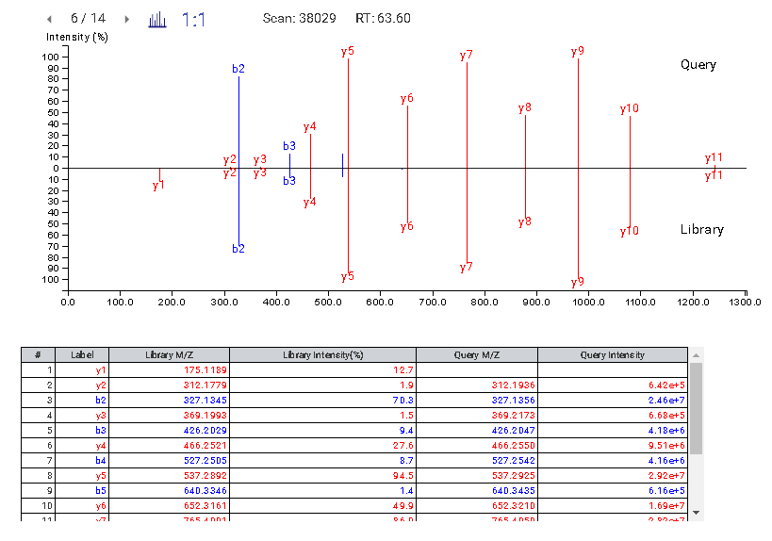
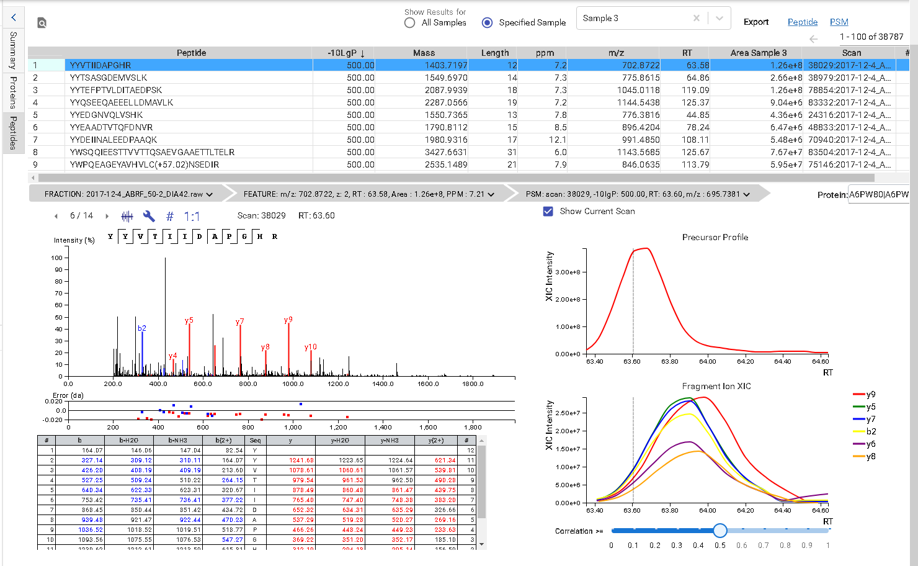
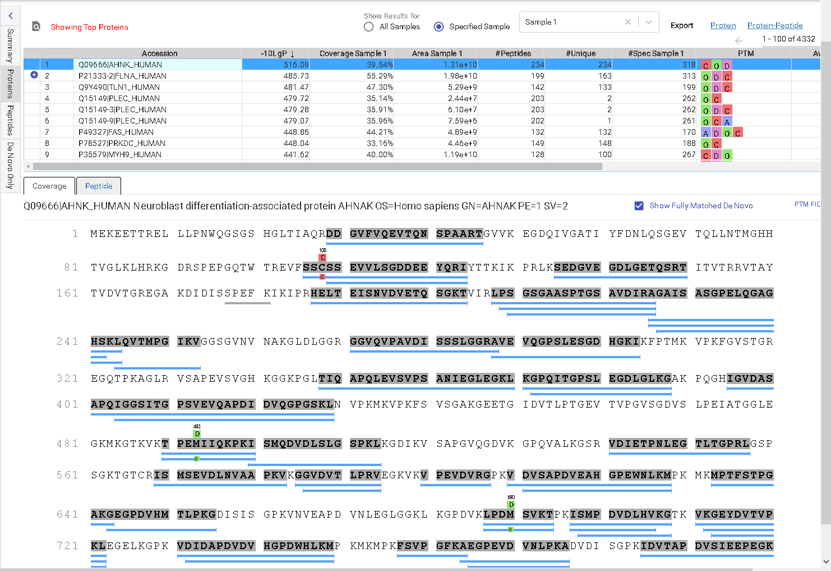
This view provides a good starting point to examine all identified peptides of a protein, with the PTMs and mutations highlighted. Clicking a peptide can bring up the peptide-spectrum matching annotation and moving the mouse over an amino acid in the annotated peptide sequence can show the corresponding supporting peaks.
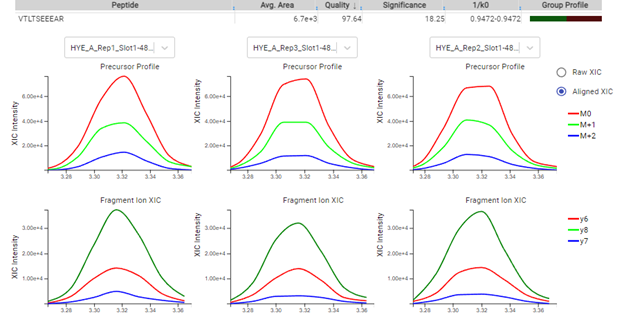
Quantify protein and peptide abundance across samples using label-free or labelled quantification techniques. Intuitive result views allow you to assess the quality of PEAKS Online sensitive and reproducible results.
PEAKS Workflow
PEAKS Online 11 is designed to facilitate accurate and sensitive proteomic analysis using the PEAKS 11 software workflows, but with higher performance on a shared resource. Users will be able to perform de novo Sequencing![]() , PEAKS DB Database Search
, PEAKS DB Database Search![]() , PEAKS Library Search
, PEAKS Library Search![]() , PEAKS PTM
, PEAKS PTM![]() , and SPIDER
, and SPIDER![]() , as in PEAKS Studio package. Optional PEAKS Q
, as in PEAKS Studio package. Optional PEAKS Q![]() , and PEAKS IMS
, and PEAKS IMS![]() modules can be enabled, for labelled and label free protein quantification and ion mobility data support.
modules can be enabled, for labelled and label free protein quantification and ion mobility data support.
PEAKS Online provides users with the ability to utilise the established PEAKS workflows more efficiently and on a larger scale. The interactive tool used to send/retrieve data to/from the server is called PEAKS Client, and the results are presented in a similar manner as available in the PEAKS Studio desktop solution. Through the Web Client Interface or Client Command Line Interface (CLI), multiple users can also access the PEAKS Online server at the same time, supporting parallelism at the project and data level.
Smart data processing, is efficient data processing. If an analysis needs to be rerun with a new set of parameters, the whole workflow does not need to be reprocessed. This is perfect for certain applications like Time Course Studies and samples can be added to an existing project and workflows can be modified “on the fly”. PEAKS Online 11 will only process the data that needs to be updated while keeping any of the relevant information generated previously. Once the desired results have been generated, users can export the results for each individual sample, or export all the results at once in a single step.
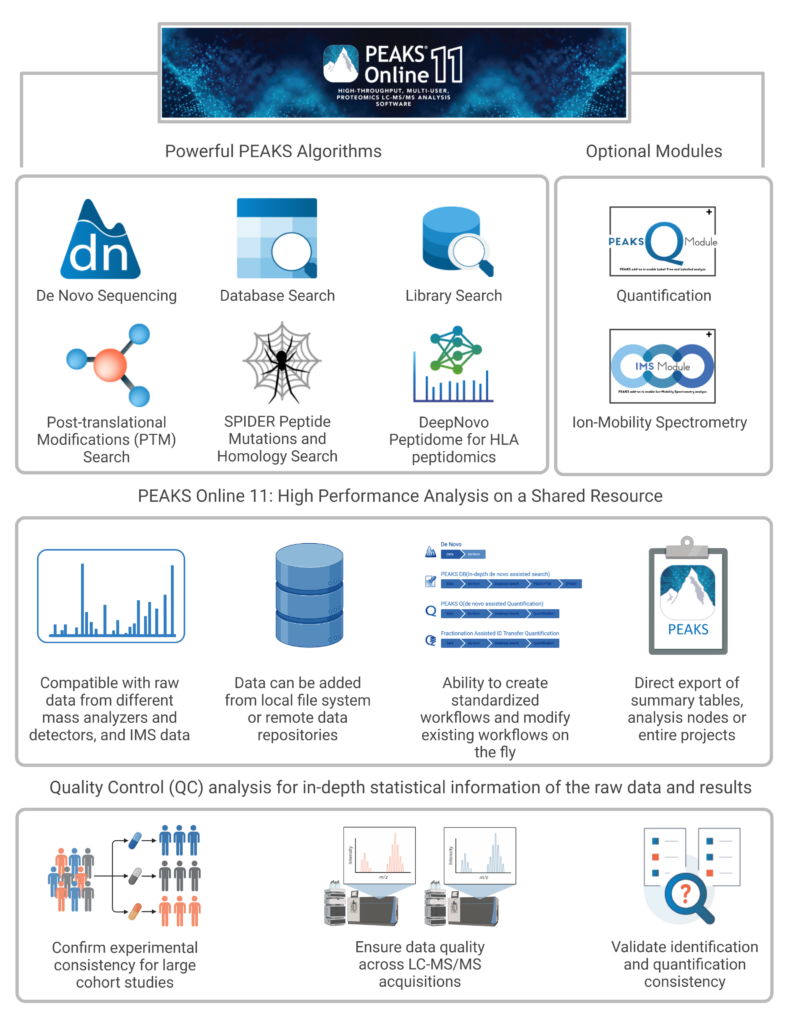
In PEAKS Online 11 dual interfaces are available to ensure easy integration into any proteomics workflow. The Web Client Interface allows users to setup and submit projects visually, as well as review and validate their results. The Client CLI on the other hand can be integrated into existing pipelines to continue automated data processing.

Web Client Interface
Access Administrative Tools
Setup, View and Share Projects
Monitor Ongoing Processes and Modify Priority
Accessible on any OS
Easily setup and access PEAKS projects from any computer, anywhere in the world. With PEAKS Online 11, users do not need to install any software files to access the server. Just open a web browser, like Google Chrome or Mozilla Firefox, to setup PEAKS searches and review the results. When your search task is done, the system will even send you an email to let you know the search is complete. Now you don’t need to worry about carrying your data around with you anymore!

Client CLI
Automated Batch Data Processing
Define Standardised Workflows
Easily Integrate into Existing Pipelines
Reduce Manual Intervention
The command line interface gives users the ability to fully automate their data analysis. Simply establish standardised workflows so that once data is created in the repository, PEAKS Online 11 will process and generate results instantaneously. Results generated using the Client CLI can be automatically exported into .csv files or viewed using the PEAKS Online 11 Web Client.
PEAKS Online 11 uses the latest technology of distributed computing to fully utilise the computing power of your hardware. The PEAKS Online 11 architecture is built on the highly popular Apache Cassandra database system, which is used in heavy-load applications like Facebook and Netflix. PEAKS Online 11 further distributes its computational workload to different workers using the Akka Actor system. This new setup gives PEAKS Online 11 the ability to do a lot of things that is was not feasible in the PEAKS Studio desktop version. In particular, PEAKS Online 11 is easily able to take on large-scale projects by more efficiently handling the increased workload of large cohort proteomics studies. In addition, its ready-to-scale, high-performance setup where the throughput and performance can be adjusted dynamically by changing the hardware configuration. Given the right resources, PEAKS Online 11 can speed up data processing at least 10 times faster than PEAKS Studio and can handle 1000 samples or more.
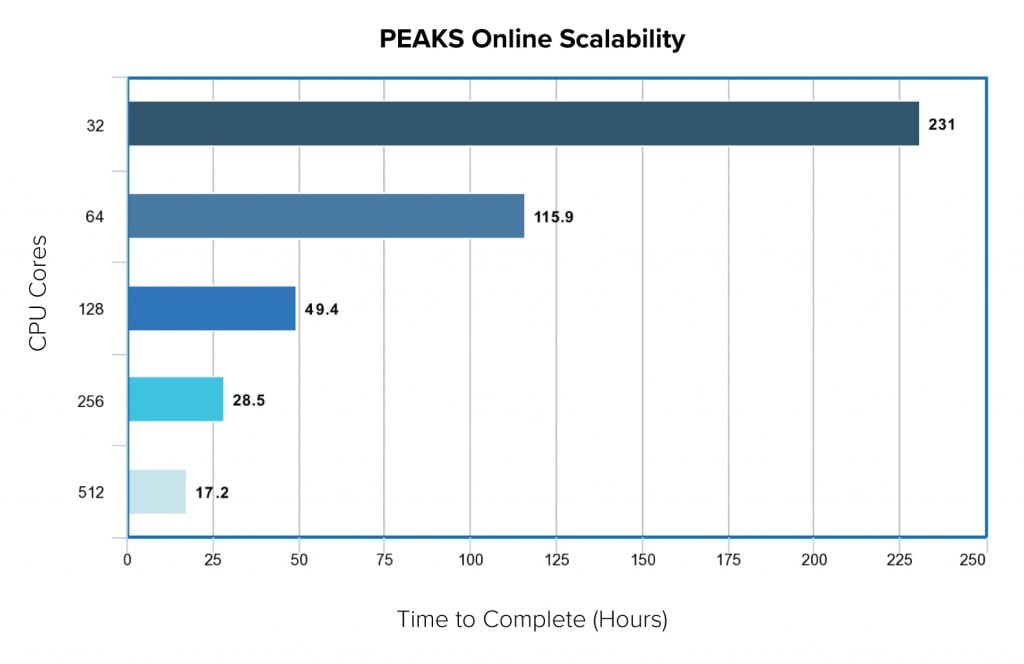
In a recent benchmarking study, we compared the scalability of PEAKS Online 11 as we increase the number of computing resources. The testing data set contained 56 samples where each sample was made up of 12 fractions. In total, we had 672 three-hour MS runs which composed of 5 million MS1 scans and 30 million MS2 scans. With the standard PEAKS Online 11 64-threads license, it took less than 5 days to finish the project, from data loading , data refinement, de novo sequencing, PEAKS DB, PEAKS PTM and SPIDER. However, when we increased the CPU cores, the performance increase linearly. With 512 cores, it took a little more than half day to finish the whole analysis.
Summary of Dataset Used for Benchmark Analysis
| # of Samples | 56 |
| # of MS/MS Runs (180 mins.run) | 672 |
| # of MS | 5106521 |
| # of MS/MS | 28858408 |
PEAKS Online is distributed as a software package that includes a Server licence with multiple Client access. The base package of PEAKS Online includes 4 Client licences and can utilise up to 64 logical cores/threads. However, PEAKS Online is ready to scale, and researchers can increase the performance and/or number of users to meet the needs of any research group. The strength of the PEAKS Online Server and number of Client licences can be purchased to align the ideal solution.
| # of Clients | # of Cores/Threads |
|---|---|
| 5 | 64 |
| 6 | 128 |
| 7 | 256 |
| 8 | 512 |
*Note: The number of cores/threads indicates the number of usable cores or threads that can be allocated to the PEAKS Online Server’s worker nodes.
Users can purchase additional individual client licences or a site client licence to meet their lab’s needs. For quantification and ion mobility data support, purchase the optional PEAKS Q and PEAKS IMS add-on respectively.
Overall, PEAKS Online 11 is composed of 3 components: the Database Node(s), Master Node, and Worker Node(s). These can be distributed to different computers, or placed on the same machine. Each component has a unique pattern of computing resource usage and hardware requirements.
Database Node(s) of PEAKS Online 11 store all application data and are the base for all proteomics processing. Since PEAKS Online 11 is a distributed computing framework that can run from multiple machines, we use the popular distributed database system Cassandra as the main data storage to provide I/O performance at scale. Each data node, as part of a Cassandra cluster, has high demand for memory space and disk I/O speed.
The Master Node is the central hub of PEAKS Online 11’s computing framework. It takes charge of scheduling, dispatching, and synchronisation of computing tasks. Although it does not perform any data processing, it takes care of the web based user interface, loading of raw data and exporting of result data, and would benefit more from high performance CPUs.
Worker Nodes are responsible for the actual data processing and computation. PEAKS Online 11 is easily scalable, so a worker node can be configured to use a customised number of CPU threads. As a rule of thumb, a worker node needs 2GB available memory for each computing thread and another 2GB spare memory for its own usage. Other than a few GB for logging, a worker node generally has no requirement for hard disk I/O speed nor space.
| 64 Thread Licence | 128 Thread Licence | |
|---|---|---|
| Master | 8 Threads, 16 GB Memory | 16 Threads, 32 GB Memory |
| Cassandra | 8 Threads Each, 16 GB Memory Each, SSD (5T+) | 8 Threads Each, 16 GB Memory Each, SSD (5T+) |
| Workers | (16x): 64 Threads Total (4 Threads Per), 160 GB Memory (10 GB Per) GPU Worker: 8 threads, 8G GPU memory, 32G system memory. | (16x): 128 Threads Total (8 Threads Per), 320 GB Memory (20 GB Per) GPU Worker: 8 threads, 24G GPU memory, 32G system memory. |
| Suggested Configuration | All-in-one Installation: INTEL XEON GOLD 6238 Processor X2 (88 Threads including hyper threading) Memory: 256 GB SSD: 500G (for OS) SSD: 5T (for Cassandra) GPU: Nvidia GTX 1080 or Nvidia Quadro P5000 16GB | Cluster with 2 machines, each has the following hardware: INTEL XEON GOLD 6238 Processor X2 (88 Threads including hyper threading) Memory: 256 GB SSD: 500G (for OS) SSD: 5T (for Cassandra) GPU: Nvidia RTX 3090 or Nvidia Quadro RTX A6000 |
Note: Although PEAKS Online 11 is best used in a cluster setting with dozens to hundreds of nodes, if you only have limited computing resources, one powerful workstation/server, you can still use it in standalone mode. In fact, all components of PEAKS Online can be started from the same machine. But in this case, you must make sure you have enough computing resources to accommodate the needs from all components. Cassandra has very intense usage of the hard drive. If everything is installed on the same machine, it’s highly recommended to have a separate high speed hard drive specifically for the data component.
References
- Lightbend, Inc. (2011-2019). Akka: Build powerful reactive, concurrent, and distributed applications more easily. https://akka.io/.
- The Apache Software Foundation (2016). Apache Cassandra. http://cassandra.apache.org/.
- Xin, L. Qiao R, Chen X, Tran NH, Pan S, Robinoviz S, Bian H, He X, Morse B, Shan B, Li M. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat. Commun. 13, 3108 (2022). https://doi.org/10.1038/s41467-022-30867-7
Resources
- PEAKS Online 11 Brochure
- Xin, L. et al. Towards Real-time Proteomics Data Analysis using PEAKS Online 8.5. ASMS. ThP 461. 7/6/2018.