Power Your Proteomics and Protein Characterisation with

We have AI-Driven Software Solutions for all your Qualitative and Quantitative Proteomics and Protein Characterisation Needs!
Connect with BSI and push the boundaries of biological research.
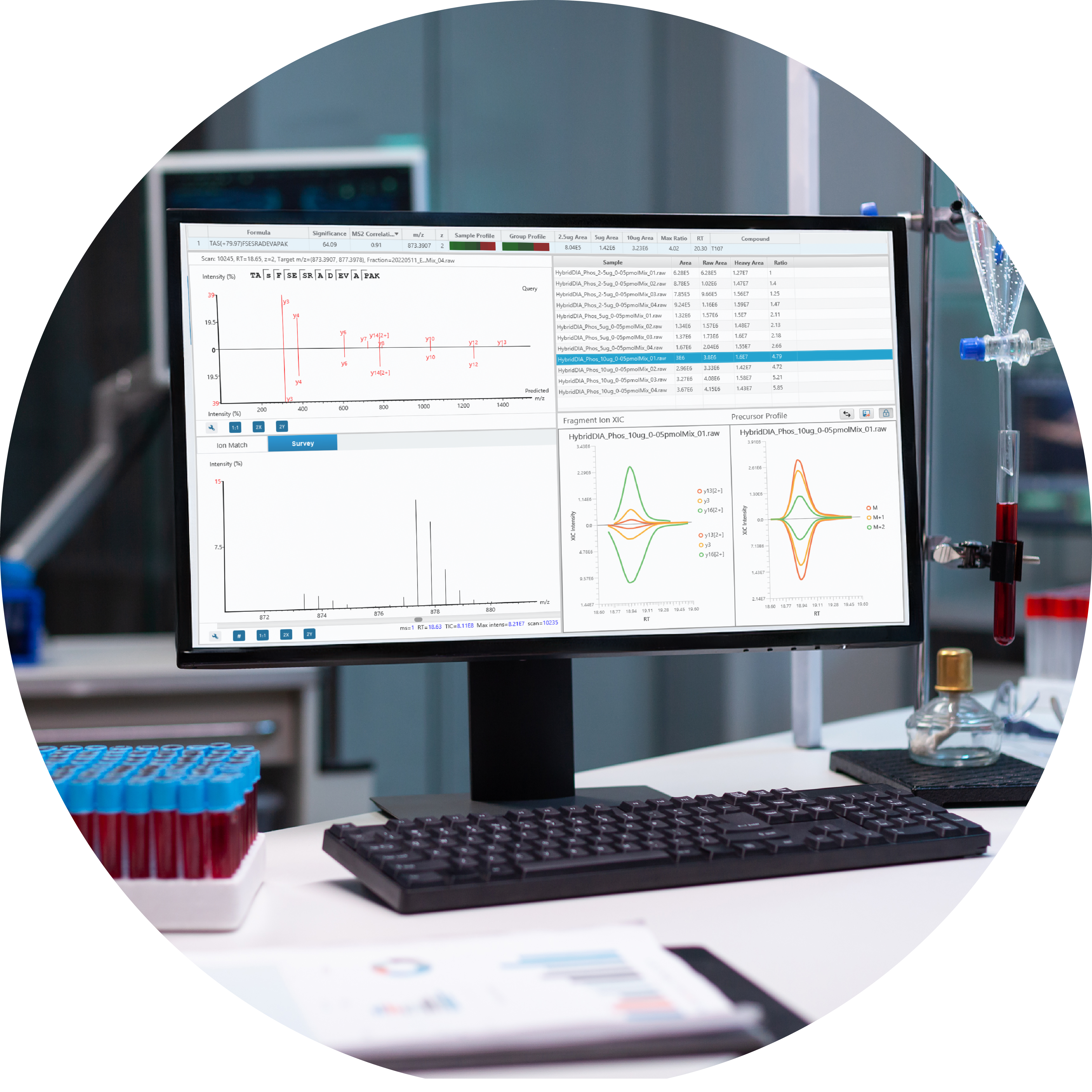

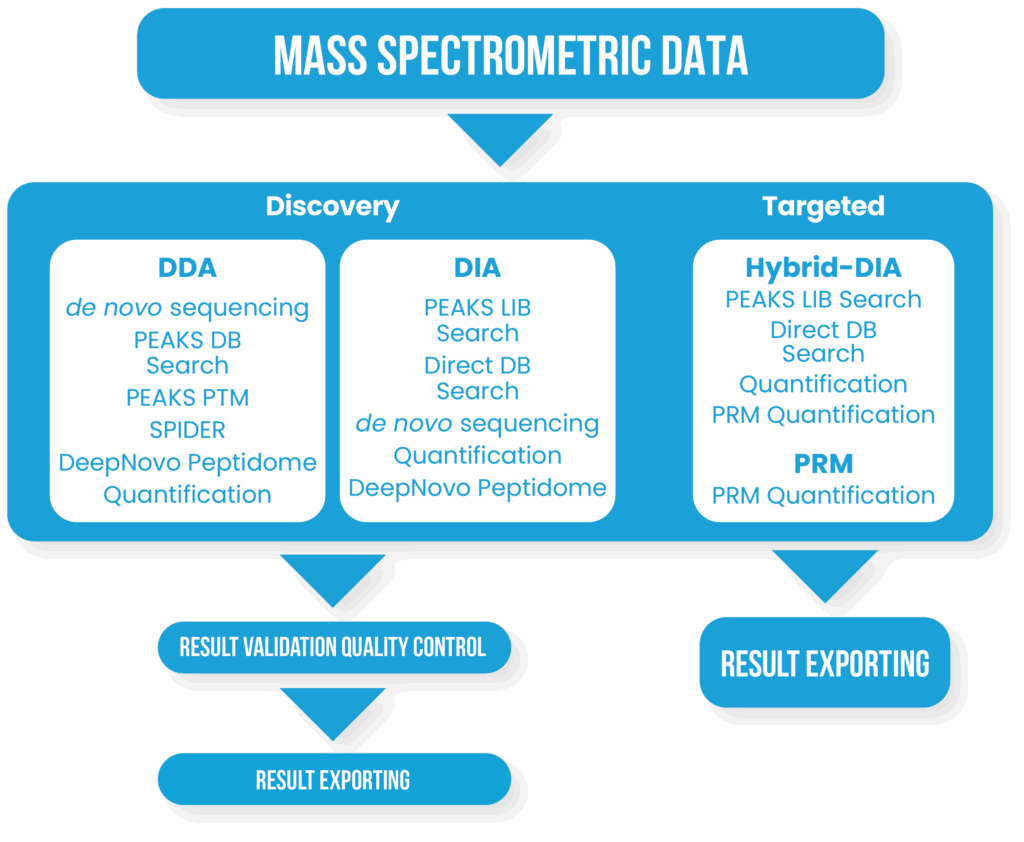
A Complete Bottom-up Discovery and Targeted Proteomics Solution
Key Highlights
- One Stop Solution for Discovery and Targeted Proteomics
- DDA and DIA Support with High Accuracy and Sensitivity
- Application Driven Solutions
- Leading de novo and DeepNovo Sequencing Powered Workflows
- Vendor Neutral
- Detailed and easy-to-use graphical user interface (GUI)

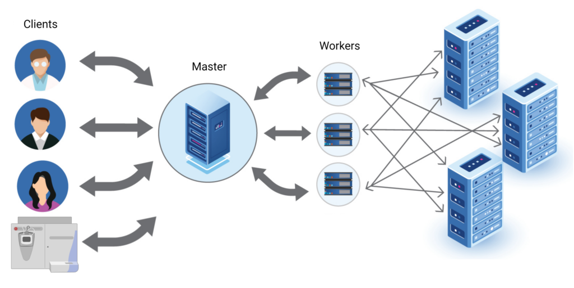
Next Level Computational Power for Large Cohort Proteomics Studies
Key Highlights
- Server-based One Stop Solution for Discovery Proteomics
- Multi-User Access for Next Level Research Demand
- Command Line Interface (CLI) for Automated Batch Submission
- DDA and DIA Support with High Accuracy and Sensitivity
- Vendor Neutral
- Detailed and easy-to-use web-based user interface

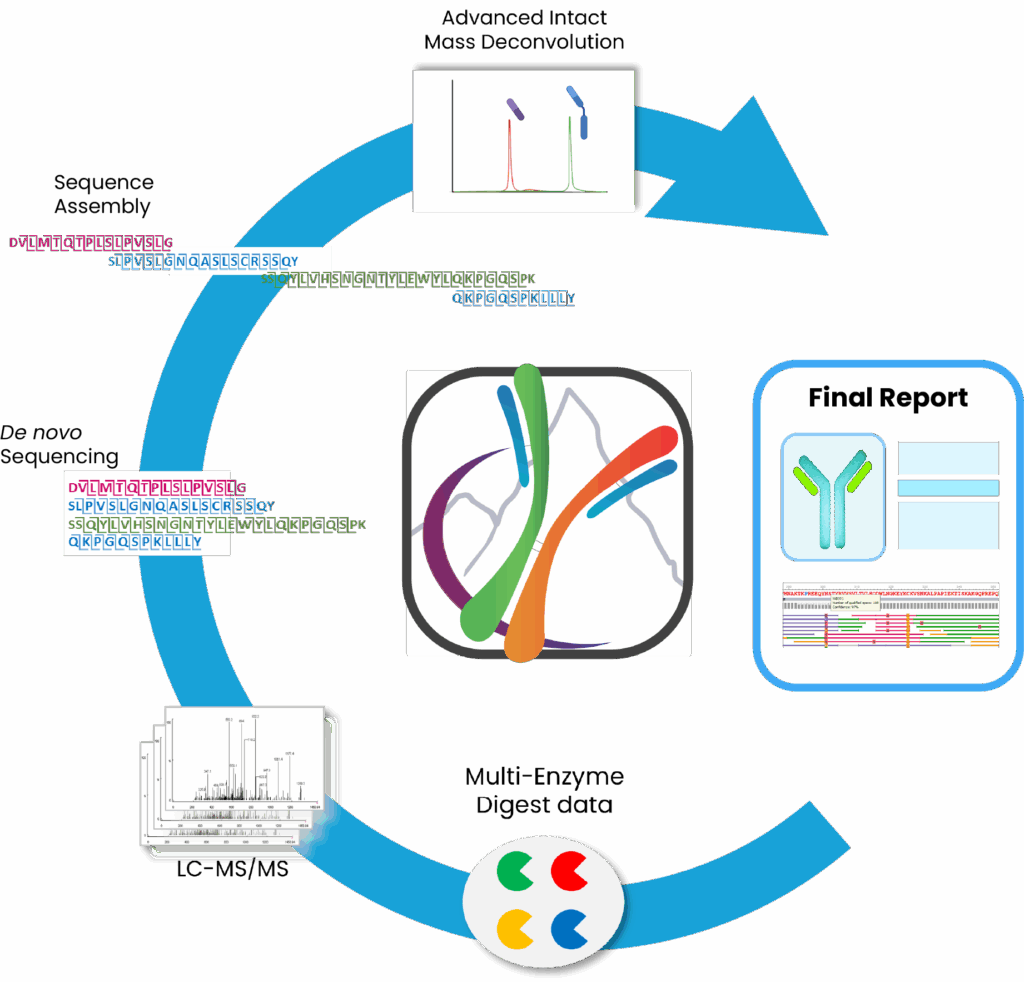
The First Automated Software Platform for Protein de novo Sequencing
Key Highlights
- Automated Protein de novo Sequencing and Protein Sequence assembly
- Integrated Advanced Bottom-up and Intact Algorithms
- In-depth Glycan profiling, PTM, and Variants Analysis
- Enhanced Ile/Leu Differentiation
- Manual Editing and Comprehensive Report Generation

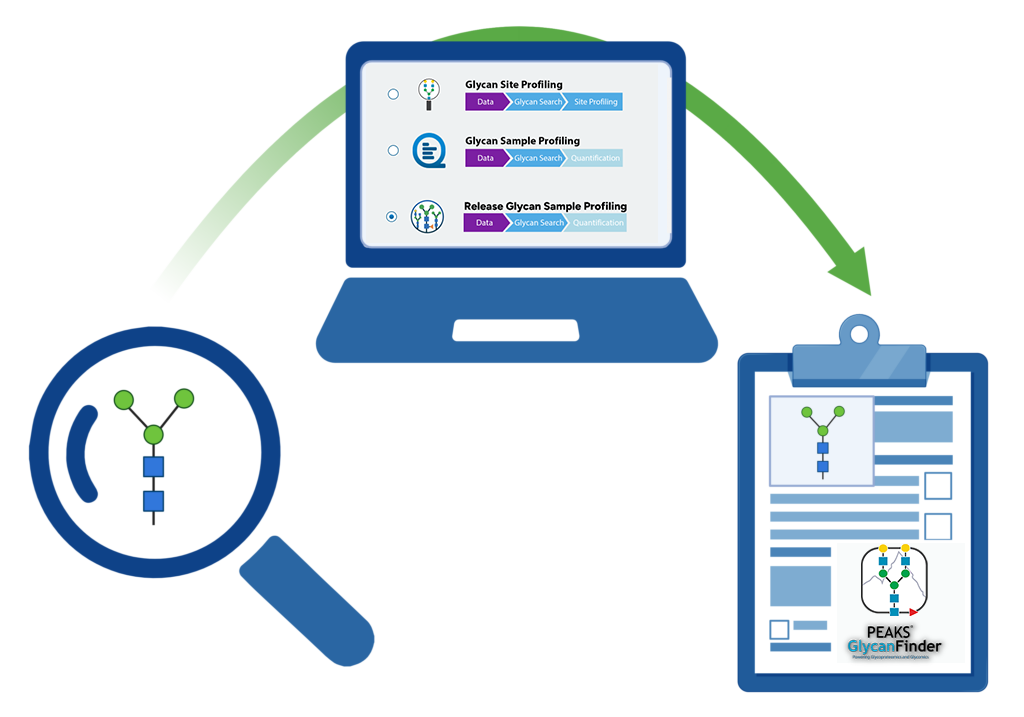
Comprehensive and Sensitive Solution for Glycopeptidomics and Glycomics with Structural Resolution
Key Highlights
- Comprehensive Glycoproteomics and Glycomics Solutions
- In-depth N- and O-linked Glycan profiling with Structural Confidence
- Accurate and Flexible Quantification for Discovery and Targeted Methodologies
- Glycan de novo Sequencing
- Vendor-Neutral
- Intuitive Visualisation and Reporting