Website builders offer a variety of templates and drag-and-drop features that enable a personalized and efficient design process. If you’re looking to craft a unique online presence, you can compare different platforms in this comprehensive comparison guide.


Proteomics is the study of proteins expressed in a given type of cell, tissue or organism under particular biological conditions at a given time. Shotgun (or bottom-up) proteomics is the most commonly used MS-based approach to study proteins by digesting proteins into peptides prior to MS analysis.
PEAKS Studio is a software platform with complete solutions for discovery proteomics, including protein identification and quantification, analysis of post-translational modifications (PTMs) and sequence variants (mutations), and peptide/protein de novo sequencing.
PEAKS Studio 11
Complete & Vendor Neutral Solution for Proteomics with DDA & DIA Support
PEAKS® Studio 11 is the next generation of the Studio platform and features a completely redesigned architecture to provide increased speed and stability. With the updated Graphical User Interface, users still get the intuitive data visualisation that PEAKS® is known for but with a new look and optimised workflows to streamline your data analysis. From DDA to DIA data support, PEAKS® Studio 11 provides a complete solution to bring your research to new heights!
Contact us to add PEAKS Studio 11 to your lab!
Highlights
- de novo sequencing.
- Database search with NEW deep learning-boost to maximise peptide ID efficiency.
- DIA workflow (Spectral Library Search + directDB + de novo).
- NEW: Direct database search using DIA data with an improved sensitivity and accuracy including DIA with short gradient.
- Enhanced Deep learning-based technology for predicting spectra, retention time, and collision cross-section values.
- Post-translational modification (PTM) search with 500+ modification.
- Sequence variant and mutation search.
- NEW: DeepNovo-based peptidome workflow for immunopeptidomics: integrated homology search with de novo for improved HLA peptidomics with FDR control.
- NEW: PEAKS Glycan module to enable a highly sensitive and accurate glycoproteomics
- Label-free and Label-based: TMT (MS2, MS3) / iTRAQ, SILAC, 18D labelling, ICAT, User-Defined.
- Quantification results can be visualised using heat maps, correlation profiles, and extracted ion chromatograms (XICs).
- Detailed and easy-to-use graphical user interface (GUI) to view, filter and validate results.
- Spectral library viewer to assess the quality and validate library before use.
- Statistical calculations presented visually to assess quality of raw data and/or results.
- LC-MS/MS heat maps provide full visualisation of peptide features, MS/MS spectra acquisition, and identification position relative to mass over charge (m/z), retention time (RT), compensation voltages (CV), ion mobility (1/k0), and signal intensity.
- NEW: PEAKS 11 now support ZenoTOF.
- Support ion mobility spectrometry proteomics (timsTOF Pro, FAIMS, HDMSe).
- Algorithms fine tuned for each instrument and fragmentation type to ensure optimal accuracy and sensitivity.
- Comprehensive support of DDA and DIA for identification and quantification analyses.
Software Overview
Algorithms
PEAKS 11 uses the latest PEAKS algorithm for all analyses, including data loading/refinement, identification and quantification. New to PEAKS Studio 11 users have the option to use Deep learning-boosted ID workflows for DDA analysis to increase of identification rate over 10%.
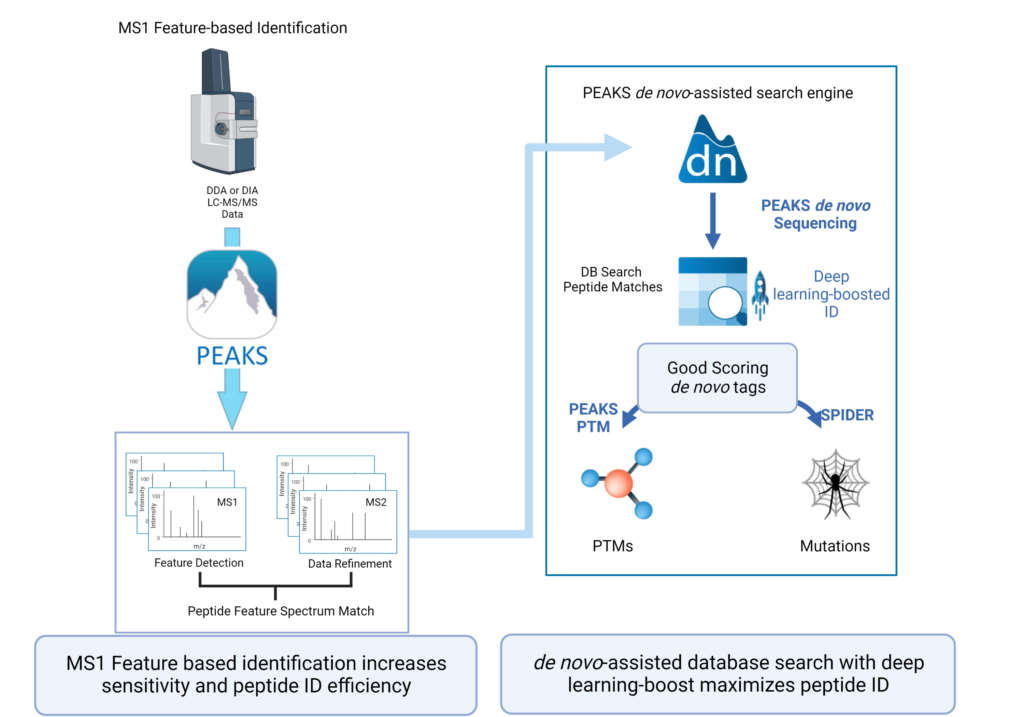
Feature-based identification workflow to increase sensitivity and maximise peptide identification efficiency.
- Designed for DDA technology to improve reproducibility.
- Integrate database search and de novo sequencing to extend in-depth analysis.
- Activate Deep learning-boost in PEAKS DDA workflows to maximise peptide ID efficiency.
Learn more about the advantages de novo sequencing brings to your research.
Streamlined Workflow with Direct Database search for DIA
DIA analysis is an appealing alternative to DDA workflows. In the past, DIA methods have relied on generating spectral libraries from DDA to identify and quantify peptides. PEAKS Studio 11 offers a unique DIA workflow to maximise identification of peptides by integrating three methods: spectral library search, direct database search, and de novo sequencing.

- A library search is performed against a predefined spectral library. Peptide spectra without a library match can be directly searched against a protein database.
- A protein sequence database is directly searched with DIA data. Advanced machine learning algorithms allow improved accuracy and sensitivity of peptide identification. During this step of the pipeline, PEAKS 11 DIA workflow now supports the identification of any PTMs specified by the user. This will enable an increase in identification of modified peptides without requiring their entries in a spectral library.
- Unmatched spectra from the database search are de novo sequenced.
- Identified peptides from both the spectrum library search and protein sequence database search can be used in a quantification analysis.
Note: All 3 search methods, PEAKS Spectral Library Search, Direct Database Search, and de novo sequencing are optional, and the steps of the workflow can be conducted independently or in sequence.
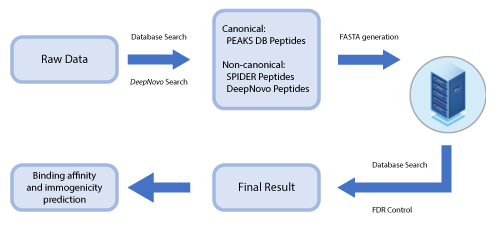
PEAKS DeepNovo Peptidome: Advanced solution Immunopeptidomics
This newly developed solution is a specialised workflow for peptidomics data that combines database searching, de novo sequencing, and identification of mutated peptides. Learn more
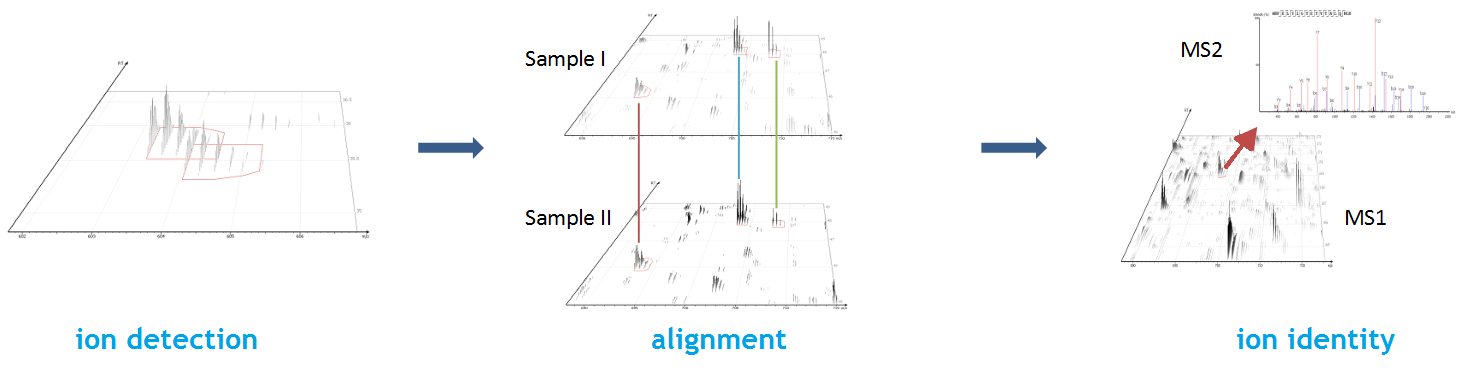
Ion mobility enabled quantification to minimise missing-values and enhance accuracy
PEAKS Glycan Module add-on enables scientists to determine glycan site localisation and glycan structures
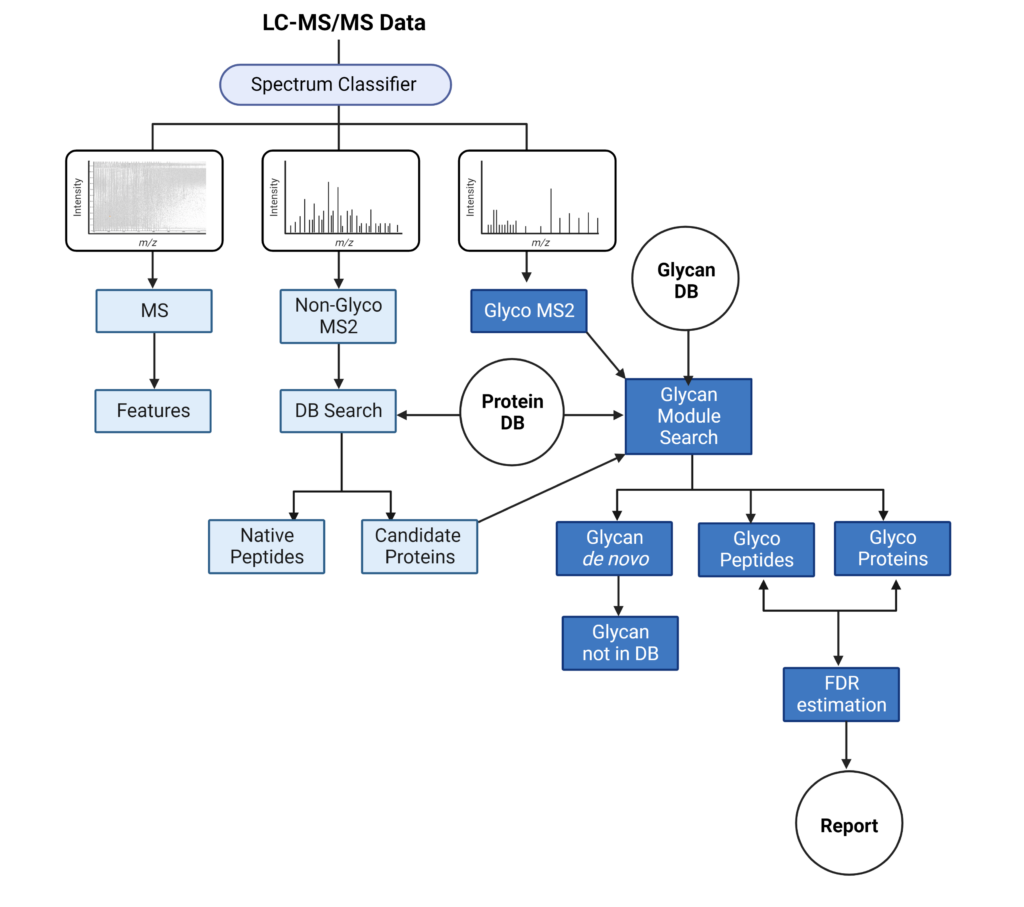
PEAKS Glycan module is a comprehensive data analysis tool that provides a highly sensitive and accurate glycoproteomics software solution to advance our understanding of the glycoproteome. Learn more
- Comprehensive understanding of glycosylation and glycoproteins
- Advanced glycoproteomic for deep glycan profiling
- Innovative software tool for glycan and glycopeptide quantification
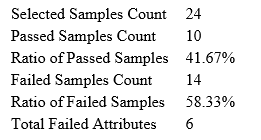
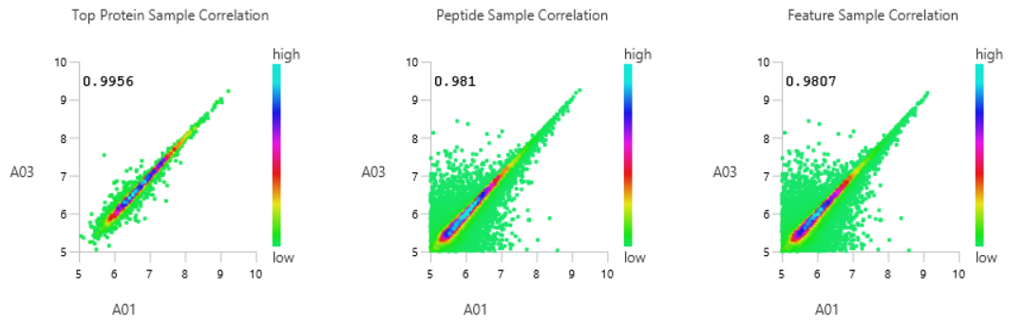
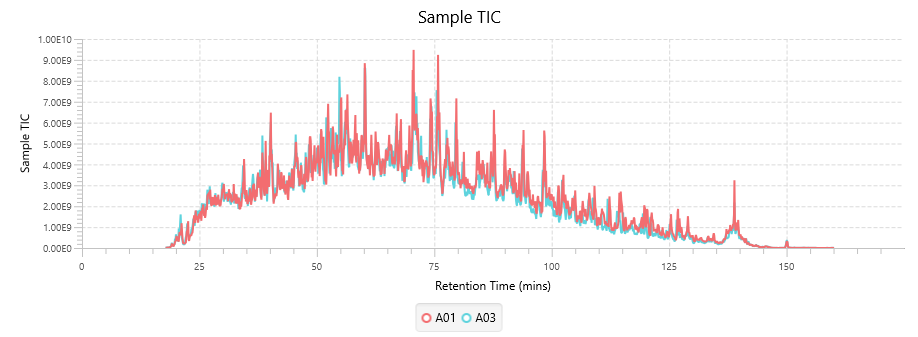
Quality Control (QC) Function for in-depth analysis from raw data to results
With the new Quality Control (QC) analysis in PEAKS® Studio 11 users can assess statistical information of the raw data and/or results and gain beneficial insight into the attributes of the LC-MS acquisition. This automated tool is designed for both, DDA and DIA data and will supply the elements to determine the quality of the data and evaluates the setup of the experiment.

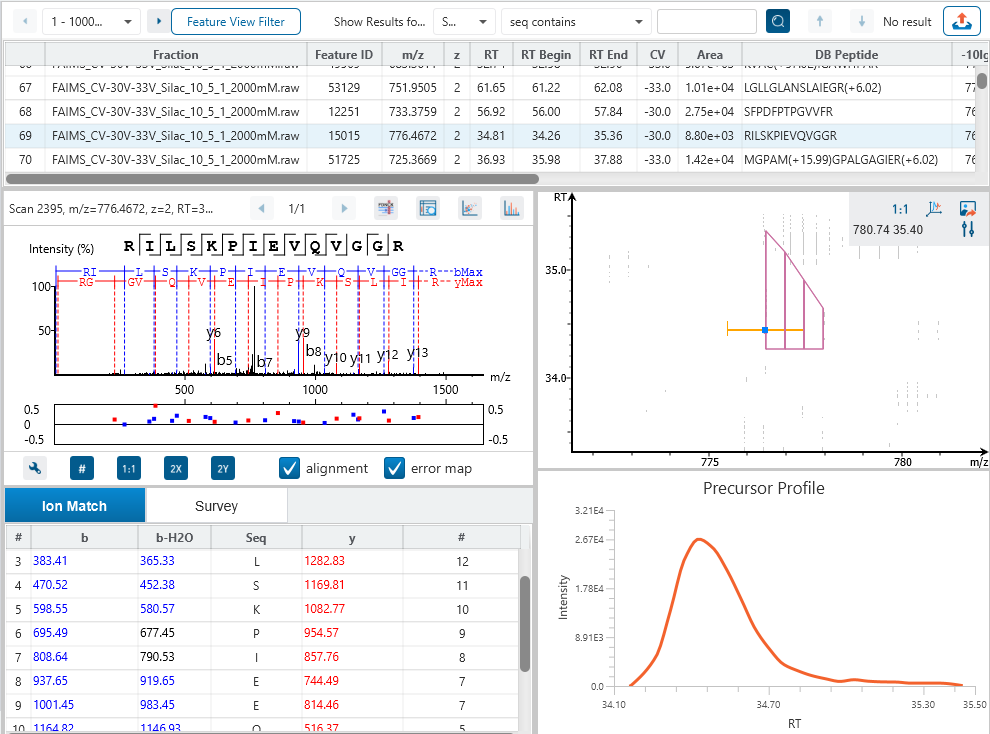
The Peptide Feature View
From data refinement to identification and quantification, the PEAKS 11 series is peptide feature-based. A peptide feature includes a series of corresponding m/z values, a retention time range, the intensities formed by different molecular isotypes, and the ion mobility as the fourth dimension information.
In PEAKS identification results, users will find a “Feature” tab which presents all details of every peptide feature in the raw data, visually and tabularly. In the Peptide Feature table, PEAKS conveniently summarises feature details detected from LC-MS or LC-IMS/MS, including the identified sequence as determined by database search, DDA, or de novo sequencing. Each feature is also linked to their corresponding protein view, spectrum view and LC-MS or LC-IMS/MS view for further inspection.
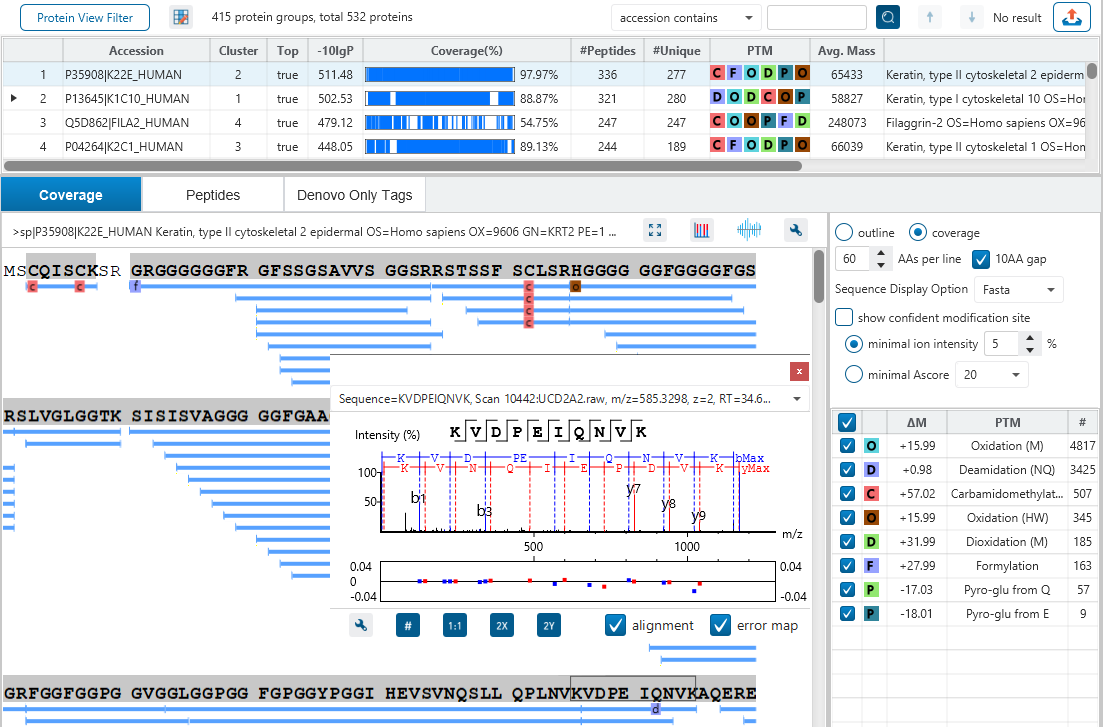
The Protein View
The Protein View presents protein profiling across complex biological samples in both DDA and DIA workflows. For each protein, the Sequence Coverage View displays a peptide map with spectrum annotation for validation. With PEAKS traditional de novo-assisted database search, users easily view identified peptide sequence in blue, while a grey bar indicates a de novo only tag match.
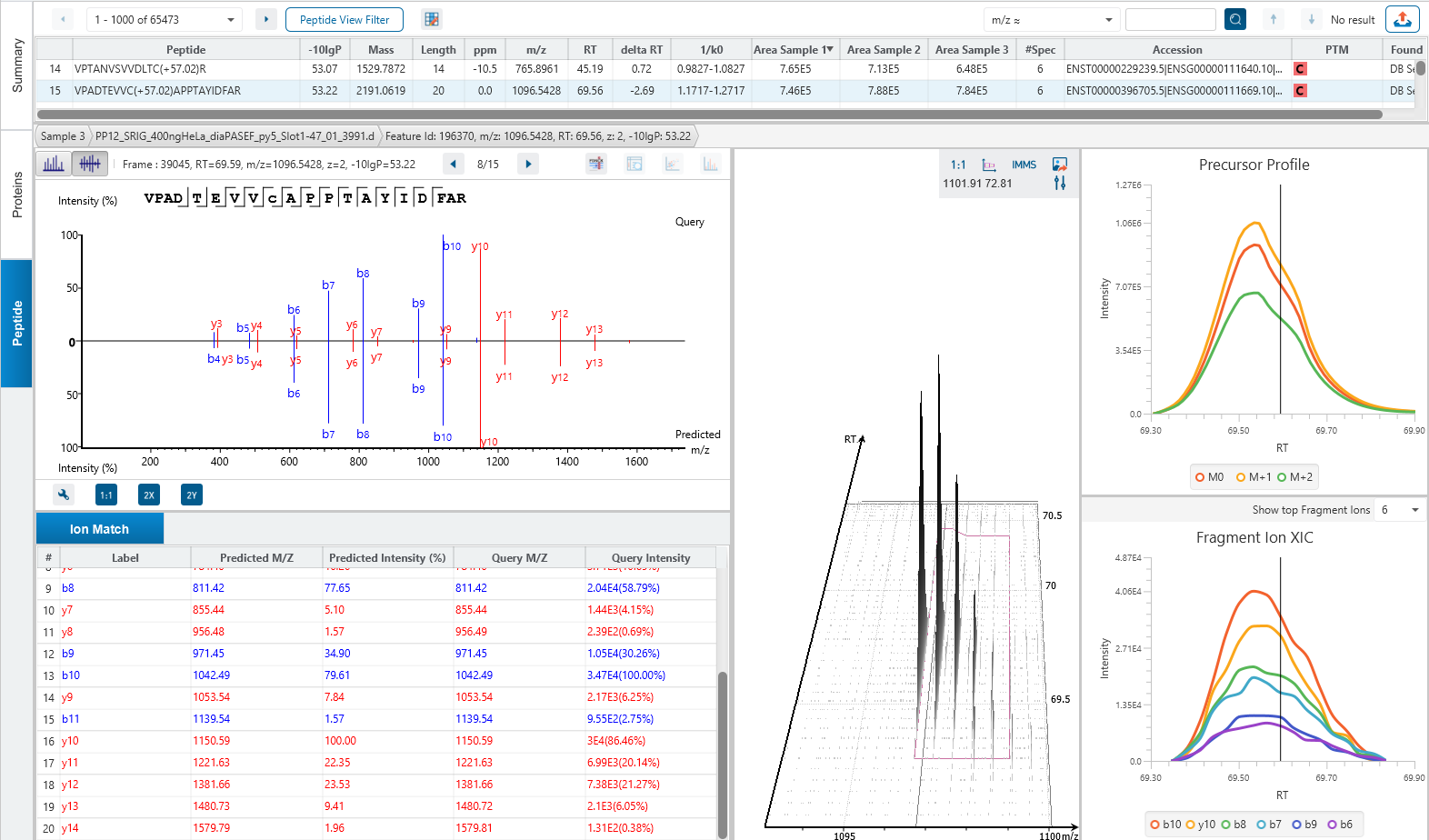
The Peptide View
The Peptide View provides a list of identified peptides with the abundance from MS1. For each modified peptide, the confidence of modification site (Ascore) is associated.
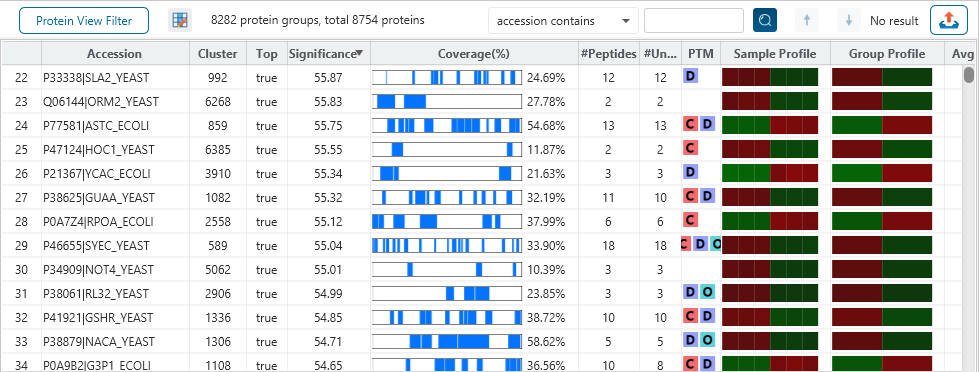
The Quantification View
With the add-on module of PEAKS Q, PEAKS Studio also determines relative protein abundance changes across a set of samples simultaneously and without the requirement for prior knowledge of the proteins involved.
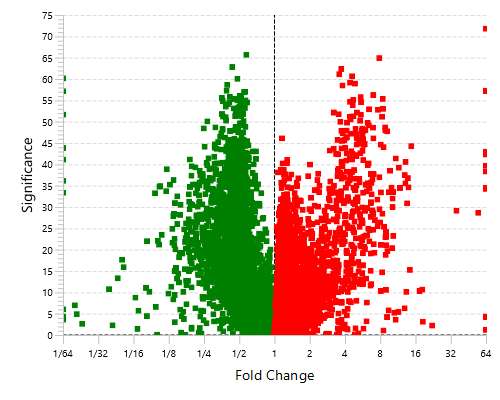
Highly differentially expressed proteins between two groups are identified by statistical analysis tool (fold change >2, FDR <0.01) and displayed in a heatmap format.
Licence Information
The PEAKS Studio licence can be scaled to address your lab’s requirements.
- Desktop – 16 threads, capable of processing across up to 16 cores
- Workstation – 32 threads, up to and across 32 cores
Configuration
PEAKS 11 is recommended to be installed on 64-bit Windows operating system with Windows 10 or later. The two main factors affecting PEAKS 11’s performance are CPU and RAM. The minimum, recommended and ideal requirements are as follows:
Supported Configuration:
- Minimum: Quad-core processors and 32 GB of RAM.
- Recommended desktop: 20 threads processors and 64GB of RAM with compatible GPU (described below).
- Recommended workstation: 40 threads processors and 128GB of RAM with compatible GPU (described below).
- Ideal: Intel Core i7/i9/Xeon or AMD Ryzen 7/9/Threadripper processors with total 32 threads or more, and 64-128 GB of RAM with compatible GPU. For running DIA Database Search, it is recommended that the machine is equipped with at least 64GB of free memory and a NVIDIA CUDA compute capability >= 4 GPU with at least 8GB of dedicated memory.
References & Resources
References
- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M, Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry, Nat. Methods, 16, 63–66 (2019). https://doi.org/10.1038/s41592-018-0260-3
- Tran NH, Zhang X, Xin L, Shan B, Li M. De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. U.S.A. 114, 8247-8252 (2017). https://doi.org/10.1073/pnas.1705691114