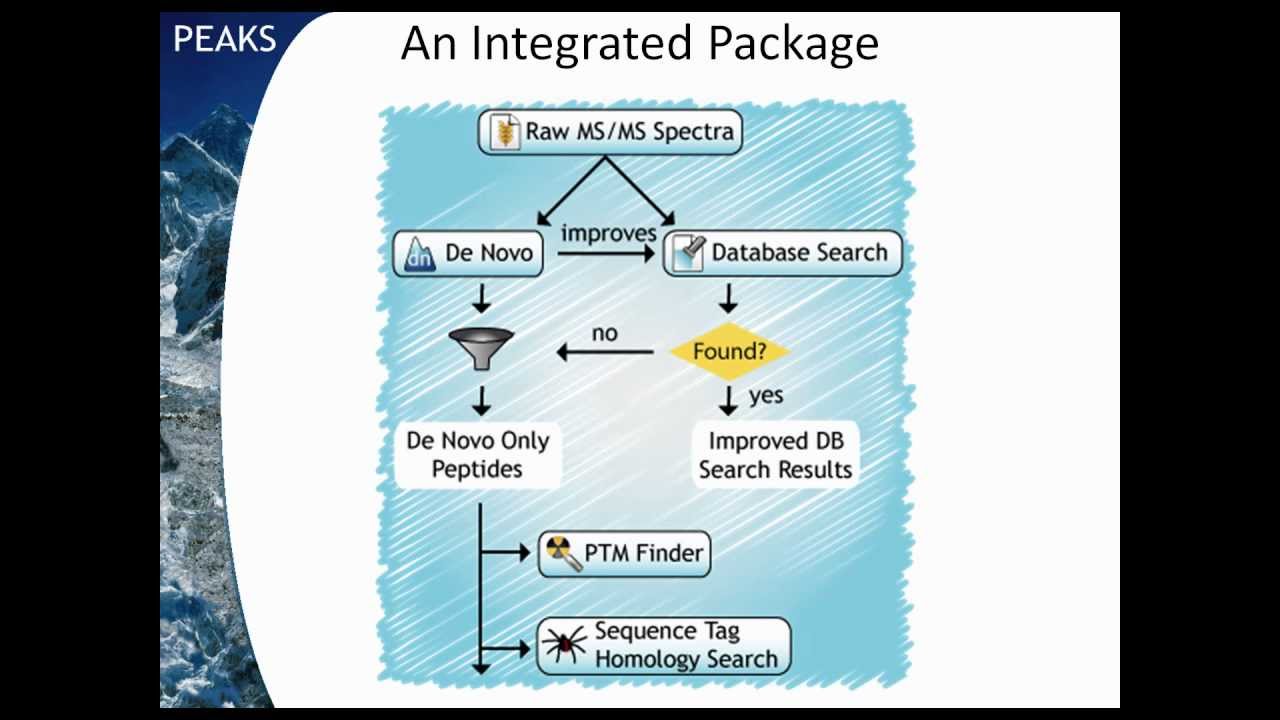
0On 2011-10-052023-03-30By Bioinformatics Solutions Inc. This video covers the de novo sequencing and database searching, which includes automated summary reporting, de novo only findings, as well as the additional features of homology searching, multi-engine reporting and quantification. Basics of PEAKS Studio 10,243 views You may also like PEAKS Studio: Protein Identification and Quantification Tutorial 859 views Basics of PEAKS Studio PEAKS Identification Walkthrough 3,397 views Basics of PEAKS Studio PEAKS Q Labelling Quantification – TMT/iTRAQ 5,256 views Basics of PEAKS Studio PEAKS Q Labelling Quantification – SILAC 2,533 views Basics of PEAKS Studio PEAKS Peptide Feature Intensities 1,512 views Basics of PEAKS Studio PEAKS Multi-Round Search 953 views Basics of PEAKS Studio PEAKS PTM Profiling 1,355 views Basics of PEAKS Studio PEAKS Q Label Free Quantification Walkthrough 3,534 views Basics of PEAKS Studio PEAKS DB Peptide Identification 6,396 views Basics of PEAKS Studio Peptide De Novo Sequencing 13,278 views Basics of PEAKS Studio









Leave a Reply
You must be logged in to post a comment.